![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
49 Cards in this Set
- Front
- Back
|
Immundefekte ・Einteilung (2) |
- primäre Immundefekte → angeboren → i.d.R. bereits im Säuglings- bzw. Kinderalter
- sekundäre Immundefekte → erworben → Auftreten im Erwachsenenalter
|
|
|
Primäre Immundefekte ・Übersicht (5) |
- Defekte der humoralen Immunabwehr ca. 50 % - Defekte der zellulären Immunabwehr ca. 10 % - kombinierte Immundefekte ca. 25 % - Phagozytendefekte ca. 15 % - Komplementdefekte < 1 % |
|
|
Primäre Immundefekte |
- Rezidivierende (antibiotikaresistent) Infektionen va im Bereich von → Mittelohr → Nasennebenhöhlen → Lunge |
|
|
Defekte der humoralen Immunabwehr (Antikörpermangelsyndrome) |
- Kongenitale Agammaglobulinämie - Selektiver IgA-Mangel - IgG-Subklassen-Defekt - Hyper-IgM-Syndrom - Common variable Immunodeficiency (CVID) |
|
|
Defekte der humoralen Immunabwehr (Antikörpermangelsyndrome) |

- machen sich i.d.R. zwischen dem 5. und 7. Lebensmonat bemerkbar, da zu diesem Zeitpunkt der mütterliche Antikörperspiegel (Nestschutz) beim Säugling sinkt - Aktive Impfungen sind bei Patienten mit humoralen Immundefekten i.d.R. sinnlos, da keine Antikörperproduktion ausgelöst werden kann (fehlender Titeranstieg). |
|
|
Kongenitale Agammaglobulinämie ・Definition (3) |
- X-chromosomale Mutation der B-Zell-spezifischen Tyrosinkinase (beteiligt an Ausreifung der Prä-B-Zellen zu reifen B-Zellen) → Mangel an B-Zellen → keine Antikörperbildung
|
|
|
Kongenitale Agammaglobulinämie |
- Säuglinge mit rezidivierenden bakteriellen Infektionen des sinobronchialen Systems und des Mittelohrs - 1/3 chronische Polyarthritis - Bei ungenügender Therapie später Infektionen der Meningen, des Gehirns und des Gastrointestinaltrakts sowie septische Krankheitsbilder |
|
|
Kongenitale Agammaglobulinämie |
- positive Familienanamnese - fehlende B-Lymphozyten - stark verminderte Konzentration aller Immunglobuline und fehlender Nachweis von Isohämagglutininen |
|
|
Kongenitale Agammaglobulinämie |
- transitorische Hypoglobulinämie des Neugeborenen |
|
|
transitorische Hypoglobulinämie des Neugeborenen ・Definition (1) |
- Nach Absinken der mütterlichen Antikörper im Blut des Neugeborenen kann die eigene Antikörperbildung bis zum 3. Lebensmonat aus bislang unbekannten Gründen verzögert sein. |
|
|
Kongenitale Agammaglobulinämie |
- lebenslange Immunglobulinsubstitution - antibiotisch Behandlung von Infekten |
|
|
Selektiver IgA-Mangel ・Definition (3) |
- häufigster primärer Immundefekt (Prävalenz 1/600) - Störung der Reifung von B-Zellen zu IgA-produzierenden Plasmazellen - 20 % familiäre Häufung |
|
|
Selektiver IgA-Mangel |
- häufig asymptomatisch (andere Immunglobuline übernehmen die Funktion des fehlenden IgA) - rezidivierenden, aber milden Atemwegs- und Darminfektionen - gehäuftes Auftreten von Autoimmunerkrankungen (Zöliakie, SLE, RA, chronisch-entzündliche Darmerkrankungen), Allergien und Malignomen |
|
|
Selektiver IgA-Mangel |
- wiederholter Nachweis einer verminderten oder fehlenden IgA-Konzentration bei normaler B-Lymphozytenanzahl |
|
|
Selektiver IgA-Mangel |
- rein symptomatisch (Antibiotika) - Cave keine intravenöse Immunglobulinsubstitution (Antikörper gegen das infundierte IgA ) (Bei Transfusionpflicht: gewaschene, leukozytendepletierte Erythrozytenkonzentrate) |
|
|
IgG-Subklassen-Defekt |
- 1/1000 - Alle 4 isotypische IgG-Subklassen können betroffen sein (IgG1- IgG4) |
|
|
IgG-Subklassen-Defekt |
- IgG2-Isotyp höchste Antikörperaktivität gegen bekapselte Bakterien → am häufigsten Symptomatisch - rezidivierenden sinobronchialen Infektionen - gehäuftes Auftreten von Autoimmunopathien (SLE, Diabetes mellitus Typ I oder ITP) |
|
|
IgG-Subklassen-Defekt |
- selektive Mangel einzelner IgG-Subklassen |
|
|
IgG-Subklassen-Defekt |
- rein symptomatisch - Bei schwerer klinischer Symptomatik können Immunglobuline substitutiert werden |
|
|
Hyper-IgM-Syndrom ・Definition (7) |
- heterogene Erkrankungsgruppe - IgA- und IgG-Mangel bei gleichzeitiger polyklonaler IgM-Erhöhung → Mutationen im Gen des CD40-Liganden auf aktivierten T-Lymphozyten → gestörte T-Zell-B-Zell-Interaktion → B-Lymphozyten erhalten kein Signal zur Produktion antigenspezifischer Immunglobuline - 70 % X-chromosomal - 30 % autosomal-rezessiv |
|
|
Hyper-IgM-Syndrom |
- rezidivierende bakterielle Infektionen im Bereich der Ohren, Nasennebenhöhlen und Atemwege - Infektionen mit opportunistischen Erregern (Pneumocystis jiroveci, Cryptosporidien, Histoplasma, Toxoplasma gondii) - Autoimmunerkrankungen (autoimmunhämolytischen Thrombopenie, Neutropenie und Anämie, Autoimmunhepatitis oder chronische Polyarthritis) |
|
|
Hyper-IgM-Syndrom |
- normale Gesamtlymphozytenzahl - erniedrigte IgA-, IgE- und IgG-Konzentration und hoher IgM-Spiegel. - Produktion von IgG-Antikörpern nach aktiver Impfung gestört |
|
|
Hyper-IgM-Syndrom |
- Immunglobuline substituieren - Bei schwerer Immunneutropenie wird G-CSF verabreicht - ggf. sollte eine Pneumocystis-jiroveci-Prophylaxe mit Cotrimoxazol erfolgen. |
|
|
Common variable Immunodeficiency (CVID) ・Definition (4) |
- variable Gruppe von Hypogammaglobulinämien → IgG-Mangel (obligat) → kombinierter IgG- und IgA-Mangel → kombinierter IgG-/IgA- und IgM-Mangel |
|
|
Common variable Immunodeficiency (CVID) |
- Erstmanifestation liegt häufig im Jugend- und frühen Erwachsenenalter (late onset) - rezidivierenden bakteriellen Infektionen im Bereich des Mittelohrs, der Nasennebenhöhlen, der Bronchien, des Lungengewebes und des Darms. - Autoimmunopathien wie der SLE, die ITP oder eine chronische Polyarthritis sind häufig - Malignomrisiko ist um das 50-Fache erhöht. |
|
|
Common variable Immunodeficiency (CVID) |
- Immunglobulinmangel - ungenügender Antikörperanstieg nach aktiver Impfung. |
|
|
Common variable Immunodeficiency (CVID) |
- lebenslange Immunglobulinsubstitution |
|
|
Defekte der zellulären Immunabwehr und kombinierte Immundefekte ・Übersicht (4) |
- DiGeorge-Syndrom - Severe combined Immunodeficiency (SCID) - Wiskott-Aldrich-Syndrom (WAS) - Ataxia teleangiectatica |
|
|
DiGeorge-Syndrom ・Steckbrief (5) |
- isolierten T-Zell-Defekt (Mikrodeletion)
- Anlagestörungen (Thymus (Hypokalzämie: Neugeborenentetanie), Epithelkörperchen, Herz und Aortenbogen (Fallot), Gesicht) - Dysmorphie: tief sitzende, abstehende Ohren, kleine Nase, kurzes Philtrum, ggf. Spaltenbildung)
- symptomatisch (Behandlung der Hypokalzämie, Infektionsprophylaxe) - T-Zell-Defekt heilt bei milden Verlaufsformen i.d.R. bis zum 2. Lebensjahr aus |
|
|
Severe combined Immunodeficiency (SCID) ・Steckbrief (4) |
- X-chromosomal oder autosomal-rezessiv Interleukin-Rezeptor-Genen 50% - Hypoplasie des Thymusgewebes - fulminante Virusinfektionen (Herpesviren), Pilzinfektionen, Diarrhö und Gedeihstörungen im Säuglings- und Kleinkindalter - Therapeutisch kommt nur eine allogene Knochenmarktransplantation in Betracht (schwerwiegendster Immundefekt) |
|
|
Wiskott-Aldrich-Syndrom (WAS) ・Steckbrief (5) |
- X-chromosomal-rezessive partielle Einschränkung der T- und B-Zell-Funktion (Mutation des Wiskott-Aldrich-Syndrom-Proteins (WASP) → eingeschränkte Zellmobilität) - klassische Trias aus Ekzem, Thrombozytopenie und rezidivierenden Infektionen - erniedrigte IgM-, normale IgG-, erhöhte IgA- und IgE-Konzentration - Therapeutisch kommt eine allogene Knochenmarktransplantation in Betracht. - Die Kinder versterben i.d.R. um das 10. Lebensjahr. |
|
|
Ataxia teleangiectatica ・Steckbrief (5) |
- autosomal-rezessive Mutationen im Gen der Proteinkinase ATN (Zellreparatur) - ab dem frühen Kindesalter Augenbewegungsstörungen, zerebellärer Ataxie, Tremor, Teleangiektasien im Bereich der Augen und der Haut und rezidivierenden Infektionen im Bereich der Atemwege. - verminderten Immunglobulinkonzentration (IgA-, IgG- und IgM-Mangel) und eine verminderte T-Lymphozyten-Zahl - Patienten Zellzerstörung vermeiden (z.B. Bestrahlungen) - versterben i.d.R. um das 30. Lebensjahr an den Folgen des malignen Lymphoms. |
|
|
Phagozytendefekte ・Übersicht (6) |
- Septische Granulomatose - Zyklische Neutropenie - Myeloperoxidasemangel - Leukozytenadhäsionsdefekt 1 (LAD1) - Chediak-Higashi-Syndrom - Shwachman-Diamond-Syndrom |
|
|
Septische Granulomatose ・Steckbrief (6) |
- häufigste Phagozytendefekt - meistens X-chromosomal, seltener autosomal-rezessiv NADPH-Oxidase Defekte (Störung des oxidativen Metabolismus von Granulo- und Monozyten) - rezidivierende akute bakterielle und mykotischen Infektionen sowie chronischen Infektionen der Lymphknoten (abszedierende Lymphadenitis), Haut, Knochen und inneren Organen mit Abszessbildung - erhöhte IgA-, IgG- und IgM-Konzentration, Leukozytose mit Neutrophilie - lebenslange Prophylaxe mit intrazellulär wirksamen Antibiotika und Antimykotika - Etwa 1/3 der Patienten stirbt im Kindesalter. |
|
|
Zyklische Neutropenie ・Steckbrief (4) |
- autosomal-dominant vererbte Mutation des Elastasegens - Etwa alle 21 Tage entwickelt sich eine Neutropenie, die etwa 1 Woche anhält - Entzündungen im Bereich der Mundhöhle und der Haut (Phlegmone) - Therapie erfolgt durch Gabe von G-CSF. |
|
|
Myeloperoxidasemangel ・Steckbrief (2) |
- häufiger Defekt, der i.d.R. asymptomatisch verläuft - Im peroxidasegefärbten Differenzialblutbild lassen sich keine neutrophilen Granulozyten nachweisen |
|
|
Leukozytenadhäsionsdefekt 1 (LAD1) ・Steckbrief (4) |
- autosomal-rezessiv β2-Integrine Adhäsionsdefekt (Leukozytenwanderung) - verzögerter Abfall der Nabelschnur. - schwere, i.d.R. ulzerierenden Hautinfektionen - Im Labor zeigt sich typischerweise eine Leukozytose. |
|
|
Chediak-Higashi-Syndrom ・Steckbrief (5) |
- autosomal-rezessiv Funktionsstörung der Granulozyten und Mangel an NK-Zellen - okulokutanen Pigmentstörung (Albinismus), sehr helle Haare, einer Hepatosplenomegalie und rezidivierende pyogene Infektionen - Granulationsanomalien (Riesengranula) in Leukozyten und Lymphozyten - allogene Knochenmarktransplantation. - Die meisten Patienten sterben um das 20. Lebensjahr. |
|
|
Shwachman-Diamond-Syndrom ・Steckbrief (2) |
- Sehr seltene, autosomal-rezessive Mutation auf Cromosom 7 - exokrine Pankreasinsuffizienz, Knochenmarkhypoplasie mit Granulozytopenie, Thrombozytopenie und Anämie |
|
|
Komplementdefekte ・Übersicht (2) |
- Mangel einzelner Komplementfaktoren - Hereditäres angioneurotisches Ödem (Quincke-Ödem) |
|
|
- Mangel einzelner Komplementfaktoren ・Steckbrief (3) |
- C5–C8-Mangel → gehäuftes Auftreten von Neisserieninfektionen (Gonorrhö, Meningitis, Sepsis) → rezidivierende pyogene Infektionen durch bekapselte Erreger (Pneumokokken, Neisserien, Haemophilus) → gehäuftes Auftreten von rheumatischen Autoimmunerkrankungen (v.a. SLE) durch eine gestörte Eliminierung apoptotischen Materials und einer gestörten Immunkomplex-Clearance. |
|
|
- Mangel einzelner Komplementfaktoren ・Therapie (1) |
・Diagnostik - Bestimmung der Konzentration der Komplementfaktoren C3/C4 (→ höchste Serumkonzentration) und der gesamthämolytische Aktivität (CH50) - Ergeben sich hierbei pathologische Ergebnisse, werden anschließend die einzelnen Komplementfaktoren untersucht.
・Therapie - Eine spezifische Behandlung gibt es nicht. Therapeutisch steht die symptomatische Therapie im Vordergrund. |
|
|
Hereditäres angioneurotisches Ödem (Quincke-Ödem) ・Definition (2) |
- Autosomal-dominant vererbter C1-Esterase-Inhibitor-Mangel - häufigster primäre Komplementdefekt |
|
|
Hereditäres angioneurotisches Ödem (Quincke-Ödem) |
- inadäquate Aktivierung des Komplementsystems → ↑Bradykinin- und Kallikreinkonzentration → ↑Gefäßpermeabilität |
|
|
Hereditäres angioneurotisches Ödem (Quincke-Ödem) |
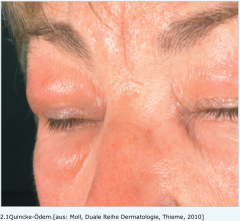
- akut auftretende, anfallsartige Schwellungen der Haut und Schleimhäute, die weder jucken noch schmerzen v.a. im Bereich des Gesichts und der Extremitäten i.d.R. zwischen 48 und 72 h - Darmkrämpfen, Diarrhö, Übelkeit und Erbrechen (Schwellung der Darmhaut) |
|
|
Hereditäres angioneurotisches Ödem (Quincke-Ödem) |
- erniedrigte Konzentration des C1-Esterase-Inhibitors |
|
|
Hereditäres angioneurotisches Ödem (Quincke-Ödem) |
- Im Anfall: intravenöse C1-Inhibitor-Substitution, alternativ auch FFP - Anfallsprophylaxe: anabole Substanzen (zb Danazol) (fördern die hepatische Synthese des C1-Esterase-Inhibitors) - ACE-Hemmer sind bei Patienten mit hereditärem angioneurotischem Ödem kontraindiziert, da sie zu einer Erhöhung des Bradykininspiegels führen und Anfälle auslösen können. |
|
|
Sekundäre Immundefekte ・Definition (2) |
- wesentlich häufiger als primäre Immundefekte und können in jedem Lebensalter - Unterernährung stellt weltweit die häufigste Ursache für einen sekundären Immundefekt dar (vor HIV). |
|
|
Sekundäre Immundefekte |
- iatrogen - Malignome - Infektionserkrankungen - Autoimmunerkrankungen - Eiweißmangelsyndrome - Malnutrition - Stoffwechselerkrankungen und metabolische Erkrankungen |