Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
133 Cards in this Set
- Front
- Back

|

|
|
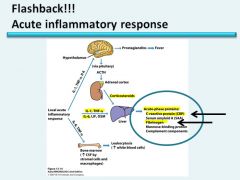
|

|
|

|

|
|
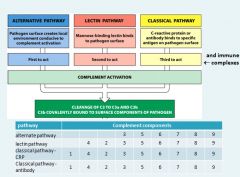
Immune complexes can fix complement and induce inflammation. When you have a disease that involves immune complexes, you may see
a decrease in the serum level of complement; because the components are being used up. |

|
|

|
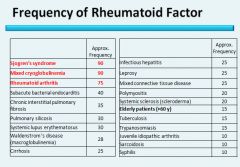
RF is non-specific
|
|

|

|
|
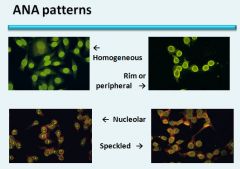
|
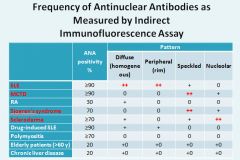
SLE: Diffuse and peripheral
MCTD: Speckled Sjogren’s: Speckled Scleroderma: Nucleolar |
|

|

|
|

|

|
|
Differential by PMN’s:
Under 50% - non-inflammatory Over 50% - inflammatory Over 90-95% - septic until proven otherwise |

Gout crystals are negatively birefringent – parallel = yellow
Tophus = a place where the body has stored crystals |
|

This crystal is not needle shaped; this is the appearance of calcium pyrophosphate dihydrate crystals = pseudogout
These crystals are positively birefringent. Not yellow when parallel |

Cholesterol crystals on the left; talcum power on the right (might see this in a patient who had recent joint surgery)
|
|
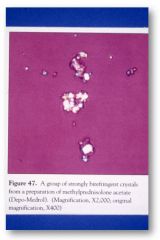
From steroid injections into a joint.
|
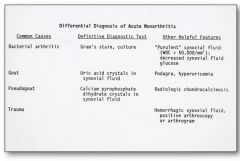
Podagra = inflammation of the great toe
KNOW THIS SLIDE |
|
|
Primary vs. Secondary Gout:
|
Primary: 90% of cases; the basic cause is unknown or when it is due to an inborn metabolic defect that causes hyperuricemia.
Secondary: 10% of cases; the cause of hyperuricemia is known, but gout is not the main clinical disorder (leukemia, renal disease, complete HGPRT deficiency - Lesch-Nyhan syndrome) |
|
|
Main morphological manifestations of gout:
|
Acute arthritis
Chronic tophaceous arthritis Tophi Gouty nephropathy |
|
|
Uric acid synthesis:
|
Uric acid is the end product of purine catabolism. The synthesis of purine nucleotides involves two different but interlinked pathways: the de novo and salvage pathways
|
|
|
The de novo pathway of uric acid synthesis:
|
Sythesizes purine nucleotides from nonpurine precursors.
The starting substrate is ribose-5-phosphate, which is converted into inosinic acid, then into either guanylic acid or adenylic acid. |
|
|
The salvage pathway of uric acid synthesis:
|
Synthesizes purine nucleotides from free purine bases, derived from dietary intake and by catabolizing nucleic acids and purine nucleotides.
In this pathway purine nucleotides are formed in a single step between PRPP and hypoxanthine, guanine or adenine. |
|
|
Lesch-Nyhan syndrome:
|
X-linked condition in which there is a complete lack of HGPRT; characterized by excessive excretion of uric acid, severe neurological disease with mental retardation and self-mutilation.
Due to absence of HGPRT, purine nucleotide sythesis via the salvage pathway is blocked. This leads to an accumulation of PRPP and increased activity of amido-PRT (from the de novo pathway). As a consequence, de novo pathway production is increased, resulting eventually in excess production of the uric acid end product. Partial deficiencies of HGPRT lead to severe gouty arthritis, occasionally with mild neurological disease. |
|
|
Diagram of uric acid synthesis:
|
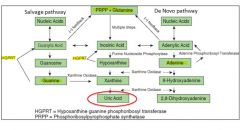
|
|
|
Pathogenesis of gouty arthritis:
|
Increased levels of uric acid lead to precipitation of monosodium urate crystals.
The crystals are chemotactic and can also activate complement to generate C3a and C5a fragments. Neutrophils and macrophages, in an attempt to phagocytose the crystals, become activated and release chemokines, toxic free radicals and leukotriene B4. The activated neutrophils also release lysomal enzymes. Macrophages secrete IL-1, IL-6 and TNF. These cytokines intensify the inflammatory response and activate the synovial and cartilage cells to release proteases - leading to tissue injury. |
|
|
Clinical course of gout:
|
1) Asymptomatic hyperuricemia
2) Acute gouty arthritis: Sudden onset of excruciating joint pain with localized erythema and warmth. The majority of first attacks are monoarticular with most in the foot or wrist. 3) Intercritital period: Patient may or may not have more attacks - usually in the absence of therapy, the attacks occur at shorter intervals and become polyarticular. After a decade or so, symptoms fail to resolve after each attack and lead to.... 4) Chronic tophaceous gout: Radiographs show characteristic juxta-articular bone erosion caused by the crystal deposits and loss of the joint space. Progression leads to severe crippling disease. |
|
|
Psuedogout:
|
Called "chondrocalcinosis" caused by calcium pyrophospate crystals.
Much of the joint pathology in psuedogout is similar to gout; with recruitment and activation of inflammatory cells. The knees, followed by the wrists, elbows, shoulders and ankles are most commonly effected. Therapy is supportive; no known treatment prevents or retards crystal formation. |
|
|
Two common causes of gout:
|
1) Decreased glomerular filtration rate
2) Diuretics * Both cause decreased excretion of uric acid. |
|
|
Rheumatoid Arthritis overview:
|
A systemic, chronic inflammatory disease affecting many tissues but mostly the joints to produce a nonsuppurative, proliferative synovitis that progresses to destroy articular cartilage and underlying bone with resultant disabling arthritis.
More common in women than men. Peak incidence is in the second to fourth decades of life. |
|
|
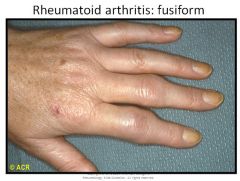
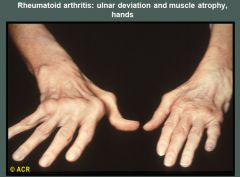
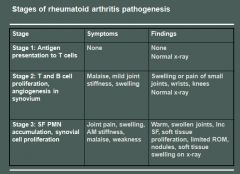
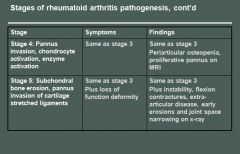
Rheumatoid arthritis morphology:
|
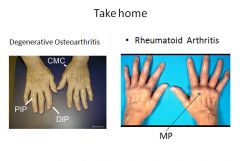
RA typically presents as symmetric arthritis, principally effecting the small joints of the hands and feet, ankles, knees, wrist, elbows and shoulders.
Typically the PIP and MCP joints are effected and the DIP are spared. Rheumatoid subcutaneous nodules develop in some patients, occuring along the extensor surface of the forearm; rarely they can form in the lungs, spleen, heart, aorta and other organs. The nodules are firm, nontender, oval or rounded masses up to 2 cm in diameter. Patients with severe arthritis, rheumatoid nodules and high RF titers are at risk for developing vasculitic syndromes. Acute necrotizing vasculitis may involve small or large arteries. Serosal involvement may manifest as fibrinous pleuritis or pericarditis. Lung parenchyma may be damaged by interstial fibrosis. Uveitis and keratoconjuctivitis may be present in some cases. |
|
|
Rheumatoid arthritis histology:
|
The effected joints show chronic synovitis, characterized by:
- Synovial cell hyperplasia and proliferation - Dense perivascular inflammatory cell infiltrates in the synovium composed of CD4+ T cells, plasma cells and macrophages - Increased vascularity due to angiogenesis - Neutrophils and aggregates of fibrin on the synovial surface and in the joint space - Increased osteoclast activity in the underlying bone leading to synovial penetreation and bone erosion. The classic appearance is that of a pannus - an overgrowth of synovial tissue that transforms the usual smooth synovial membrane into a lush, frondlike membrane. With full blown inflammatory joint involvement, soft tissue edema develops leading to fusiform swelling of the PIP joints. With progression, the articular cartilage under the pannus is eroded and destroyed. The bone may also be attacked and eroded. Eventually the pannus fills the joint space and fibrosis and calcification may cause permanent ankylosis. |
|
|
Rheumatoid Arthritis pathogenesis:
|
RA is immunologically mediated with genetic predisposition; but the exact mechanisms are unclear.
Theoretically: 1) CD4+T cells are activated somehow - leading to activation of macrophages and inflammatory response. 2) B cells are activated and produce antibody 3) Cytokines stimulate proliferation of synovial cells and fibroblasts. They also stimulate secretion of MMP 4) T cells express RANK ligand, which induces osteoclast differentiation and activation 5) RF antibodies (IgM to the Fc portion of IgG) form immune complexes with self-IgG that deposit in joints and tissue leading to inflammation and joint damage * HLA-D4 and PTPN22 genes are associated with RA * TNF plays the most important role in disease progression |
|
|
Rheumatoid arthritis clinical course:
|
Along with symmetrical polyarticular arthritis - may also see constitutional symptoms such as weakness, malaise and low grade fever due to IL-1 and TNF.
RA starts with morning stiffness and aching of the joints. As RA advances, the joints become enlarged and motion is limited. Vasulitic involvement of the extremities may lead to Raynaud phenomenon and chronic leg ulcers. To make correct diagnosis: - Characteristic radiology findings - Sterile, turbid synovial fluid with inclusion bearing neutrophils - RF |
|
|
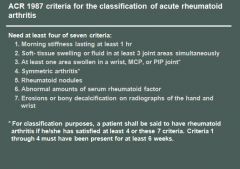
What autoantibody is highly specific for rheumatoid arthritis?
|
Anti-CCP
|
|
|
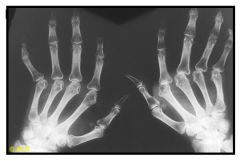
Rheumatoid arthritis: hands, advanced deformity.
The metacarpophalangeal joints demonstrate marked narrowing, subluxation, and ulnar deviation. Erosions are seen in the metacarpal heads. The proximal interphalangeal joints are narrowed, but there is no reactive bone change. Demineralization is present in bones adjacent to the metacarpophalangeal and proximal interphalangeal joints. The carpal spaces are narrowed, and erosions are present in the carpal bones and ulnar styloid processes. |
|
|
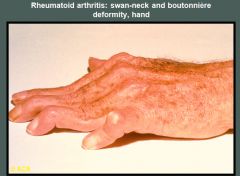
Swan-neck deformities are seen in the second, third, and fourth digits of a patient with chronic rheumatoid arthritis. A boutonnière deformity of the fifth digit is present.
|
|
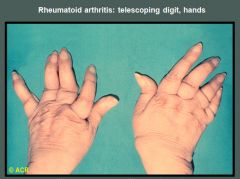
This patient with longstanding rheumatoid arthritis demonstrates the "opera-glass hand" or main en lorgnette deformity, characterized by severe joint destruction, loss of bone substance, and shortening of the digits. These changes have resulted in telescoping and hypermobility of the finger joints. The proximal interphalangeal joints and some of the metacarpophalangeal articulations are most severely involved.
|
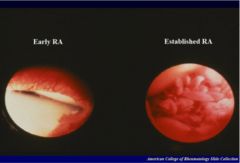
Intense inflammatory and increased vascularization in early RA
Pannus in late RA |
|
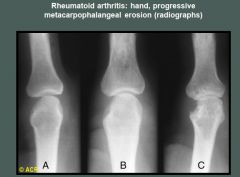
|
|
|
|
|
|
|
Felty's syndrome (a complication of RA):
|
Felty's syndrome is defined by the presence of three conditions: rheumatoid arthritis, an enlarged spleen (splenomegaly), and an abnormally low white blood count. It affects less than 1% of patients with rheumatoid arthritis.
The symptoms of Felty's syndrome are similar to those of rheumatoid arthritis. Patients suffer from painful, stiff, and swollen joints, most commonly in the joints of the hands, feet, and arms. In some affected individuals, Felty's syndrome may develop during a period when the symptoms and physical findings associated with rheumatoid arthritis have subsided or are not present. In this case, Felty's syndrome may remain undiagnosed. In more rare instances, the development of Felty's syndrome may precede the development of the symptoms and physical findings associated with rheumatoid arthritis. Felty's syndrome is also characterized by an abnormally enlarged spleen (splenomegaly) and abnormally low levels of certain white blood cells (neutropenia). As a result of neutropenia, affected individuals are increasingly susceptible to certain infections. Individuals with Felty's syndrome may also experience fever, weight loss, and/or fatigue. In some cases, affected individuals may have discoloration of the skin, particularly of the leg (abnormal brown pigmentation), sores (ulcers) on the lower leg, and/or an abnormally large liver (hepatomegaly). In addition, affected individuals may have abnormally low levels of circulating red blood cells (anemia), a decrease in circulating blood platelets that assist in blood clotting functions (thrombocytopenia), and/or inflammation of the blood vessels (vasculitis). |
|
|
Adult onset Still's disease (a complication of RA):
|
Adult-onset Still's disease is a rare form of inflammatory arthritis that was characterized by EG Bywaters in 1971. The underlying cause is unknown. It usually presents with high spiking fevers, joint and muscle pains, a salmon colored rash and other symptoms of systemic inflammation.
The disease typically affects 16–35 year olds and presents with arthralgia, elevated serum ferritin, a 'salmon-pink' rash, pyrexia and lymphadenopathy. Rheumatoid factor (RF) and anti-nuclear antibody (ANA) are classically negative. Patients experiencing a flare-up from Adult-onset Still's disease usually report extreme fatigue, swelling of the lymph glands, and less commonly fluid accumulation in the lungs and heart. |
|

|
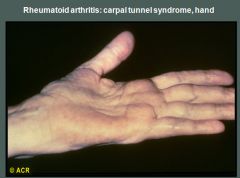
Compression of the median nerve in the carpal tunnel is not an unusual finding in patients with rheumatoid arthritis, although it may also occur in other disorders. In this case, thenar atrophy has resulted from damage to the median nerve caused by pressure from swollen inflammatory tissues. The volar aspect of the wrist is swollen. The characteristic sensory symptoms (usually involving the first three digits and radial side of the fourth digit) may be elicited by tapping over the median nerve in the volar aspect of the wrist (Tinel's sign) or by flexing the wrist (Phalen's sign).
|
|

|

|
|

|

|
|
|
Age distributions of infectious arthritis:
|
Under 2 yo - H. influenza
Older children and adults - S. aureus Late adolescence and young adult - N. gonorrhoeae People w/sickle cell at any age - Salmonella |
|
|
The seronegative spondyloarthropathies:
|
A group of diseases that develop in genetically susceptible individuals and are triggered by environmental/infectious agents.
The symptoms are immune mediated and triggered by a T cell response against an antigen that cross reacts with molecules of the MSK system. These diseases include: anklyosing spondylitis, reactive arthritis (Reiter syndrome), psoriatic arthritis and arthritis associated with IBD. Many are associated with the HLA-B27 allele and a triggering infection but without specific antibodies. * Characterized by pathological changes that begin in the ligamentous attachments to bone rather than in the synovium* |
|
|
Ankylosing Spondyloarthritis:
|
A chronic synovitis that causes destruction of articular cartilage with resultant bony ankylosis of the sacroiliac and apophyseal joints.
Inflammation of tendinoligamentous insertion sites leads to ossification, producing squaring and fusion of the vertebral bodies and bony outgrowths, which results in spinal immobility. Usually occurs between 2nd and 3rd decade of life and more often in men than women. Fracture of the spine, uveitis, aortitis and amyloidosis are other recognized complications. |
|
|
Reiter syndrome:
|
A form of reactive arthritis that is defined by a triad of arthritis, nongonococcal urethritis or cervicitis and conjunctivitis.
Most affected are men in thier 20's or 30's. This disease is caused by autoimmune reaction initiated by prior infection of the GI tract (Shigella, salmonella, yersinia, campylobacter) or GU system (Chlamydia). Symptoms develop within weeks of the inciting bout of urethritis or diarrhea. Joint stiffness and lower back pain are common early symptoms. The ankles, knees and feet are effected most often in an asymmetrical pattern. Extra-articular manifestations are inflammatory balanitis, conjunctivitis, cardiac conduction abnormalities and aortic regurgitation. |
|
|
Enteritis associated arthritis:
|
Caused by GI infection by yersinia, salmonella, shigella and campylobacter.
The LPS layer stimulates the immune response. The arthritis appears abruptly and tends to involve the knees and ankles but sometimes the wrists, fingers and toes. It lasts for about a year, then generally clears on its own. |
|
|
Psoriatic arthritis:
|
A chronic inflammatory arthritis that develops in more than 10% of the psoriatic population.
Symptoms may manifest acutely or have a long onset. The DIP joints of the hands and feet are first affected in an asymmetric distribution. The large joints of the ankles, knees, hips and wrist may be involved as well. Extra-articular manifestions are uveitis and iritis. Similar to RA - but usually not as severe, joint destruction is less frequent |
|
|
Is back pain caused by ankylosing spondylitis improved by exercise?
|
YES. It starts as morning stiffness that is improved by exercise.
|
|
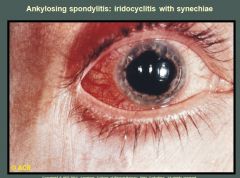
Ciliary injection and irregularity of the pupil are present. Adhesions between the iris and lens (synechiae) appear at the 1-o'clock and 4-o'clock positions. Synechiae are caused by repeated episodes of iritis and may eventually lead to glaucoma and blindness. The pupil is dilated from medication. The medial aspect of the sclera has been blanched by topical medication. Synechiae is the adhesion of the iris to the lens behind.
|
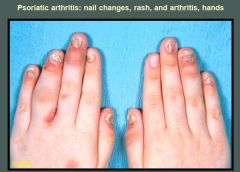
Characteristic erythema and swelling of the distal interphalangeal joints are displayed in both hands. In addition, the left fourth proximal interphalangeal joint is diffusely swollen, giving a "sausage-like" appearance. The typical nail changes of psoriasis include fragmentation, discoloration, pitting, and onycholysis (distal separation of the nail from its base due to subungual hyperkeratosis). Psoriatic skin changes are seen at the base of the fourth finger and dorsum of the left hand.
|
|
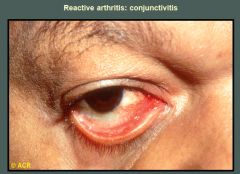
The erythema and exudate on the bulbar and palpebral conjunctivae are characteristic of the acute transient conjunctivitis of reactive arthritis. The reaction is often mild and easily overlooked, but photophobia, excessive lacrimation, burning, and intense hyperemia may occur. Iritis and episcleritis usually do not involve the palpebral conjunctivae.
|
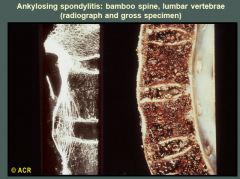
The radiograph of the lumbar spine on the left and the sagittal section (anterior elements) on the right demonstrate bony bridging of adjacent vertebrae as a result of ossification in the peripheral portions of the intervertebral discs (annulus fibrosus). This change is most severe anteriorly (see radiograph) and results in the so-called bamboo spine.
|
|
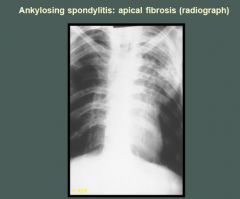
Linear and patchy infiltrates with cavity formation are present in the left upper lobe of this patient with ankylosing spondylitis. Similar but less marked infiltrates without cavitation are located in the right apex.
|

Fusiform soft-tissue swelling involves the index and middle fingers with narrowing of all the interphalangeal and metacarpophalangeal joints. Resorption of the terminal tuft of the index finger and thumb is seen. Subchondral bone resorption of the distal interphalangeal joint of the thumb and middle fingers has resulted in the "pencil-in-cup" appearance. A flexion deformity of the distal interphalangeal joint of the small finger is present, and the corresponding joint of the ring finger has fused.
|
|
|
What types of treatments are there for ankylosing spondylitis or psoriatic arthritis?
|
Treatments are strong NSAID’s, muscle relaxers, anti-TNF, stretching and yoga type exercises.
|
|
|
SLE overview:
|
SLE is a multisystem autoimmune disease of many manifestations and variable behavior.
It principally affects the skin, kidney, serosal membranes, joints and heart. It is associated with a wide variety of autoimmune antibodies, including ANA's. SLE is fairly common; it affects black American women most often. The usual onset is in the 2nd to 3rd decade of life. |
|
|
SLE etiology:
|
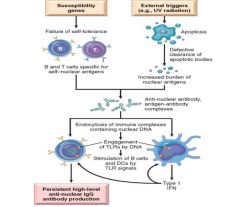
The fundamental defect in SLE is a failure to maintain self-tolerance.
The most frequently associated autoantibodies in patients with SLE are anti-dsDNA and anti-Sm. One model for the pathogenesis of SLE proposes a combination of increased generation or defective clearance of nuclear antigens released from apoptotic cells, and a failure of B and T cell tolerance to these self antigens. There are many genetic variables that can predispose to development of SLE; family history, HLA-DQ allele and C1q complement component deficiency. Non-genetic variables include; UV exposure, procainamide and hydralazine ingestion. |
|
|
SLE pathogenesis:
|
Auto-antibodies are the mediator of tissue injury. Most of the systemic lesions are mediated by immune complexes (type III).
DNA/anti-DNA complexes can be detected in the glomeruli - in addition, autoantibodies against red cells, white cells and platelets promote destruction and phagocytosis of these cells (type II). In tissues, nuclei of damaged cells react with ANA's, lose their chromatin pattern and become homogenous, to produce LE bodies or hematoxylin bodies. |
|
|
SLE organ involvement:
|
Acute necrotizing vasculitis: effects small arteries and arterioles and may be present in any tissue. In chronic stages, vessels show fibrous thickening with luminal narrowing.
Kidney: The pathogenesis of all forms of glomerulonephritis in SLE invovles DNA/anti-DNA complex deposition in the glomeruli. This leads to: mesangial lupus GN, focal proliferative GN, diffuse proliferative GN (most serious and most common in SLE) and membranous GN. Skin: Butterfly rash, UV light exacerbates the erythema - a similar rash may be present elsewhere, frequently in sun exposed areas. There is liquifactive degeneration of the basal layer of the epidermis, edema at the dermoepidermal junction and mononuclear infiltrates around blood vessels and skin appendages. IF reveals deposition of Ig and complement at the DE junction. Joints: When present, it consists of swelling and a nonspecific infiltration of mononuclear cells in the synovial membranes. CNS: Focal neurological deficits and neuropsychiatric symptoms are common; often ascribed to vascular lesions causing ischemia or multi-focal cerebral microinfarcts. Serosal membranes: The pericardium and pleura may show inflammatory changes ranging from effusions to fibrinous exudates to fibrosis. Heart: Valvular nonbacterial verrucous endocarditis (Libman-Sacks) takes the form of irregular 1-3mm warty deposits on either side of the leaflets. Other: Spleen enlargement, interstitial lung fibrosis, inflammation of liver portal tracts |
|
|
Clinical manifestations of SLE:
|
Classic presentation is a young woman with a butterfly rash, fever, arthritis, pleuritic chest pain and photosensitivity. However, most cases do not have this presentation.
The course of SLE is variable. Can progress slowly or rapidly - can go into remission for long periods of time. Renal failure, intercurrent infections and diffuse CNS involvement are the major causes of death in SLE. |
|
|
Criteria for diagnosis of SLE:
|
A person is said to have SLE if 4 of the following 11 are present.
1) Malar rash (butterfly) 2) Discoid rash 3) Photosensitivity 4) Oral ulcers 5) Arthritis 6) Serositis 7) Renal disorder 8) Neuro disorder (seizures or psychosis) 9) Hematologic disorder 10) Immunologic disorder 11) ANA |
|

|

|
|

|

|
|

|

|
|
|

|
|
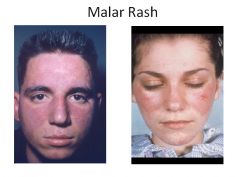
Sparing of the nasolabial folds is characteristic of a malar rash
|
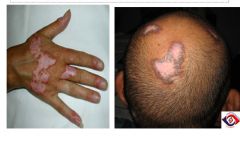
Discoid Lupus; you can have discoid lupus all by itself, or as part of systemic SLE
|
|
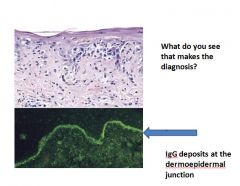
A, An H&E-stained section shows liquefactive degeneration of the basal layer of the epidermis and edema at the dermoepidermal junction. B, An immunofluorescence micrograph stained for IgG reveals deposits of Ig along the dermoepidermal junction.
|
|
|

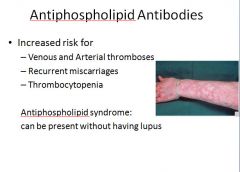
MOA: react with protein/phospholipid complex on cell membranes making it prothrombotic then bind to anticoagulants
Factor XII or Protein C Can cause a false positive with a syphilis test |
|
|
|

|
|

|

|
|
|
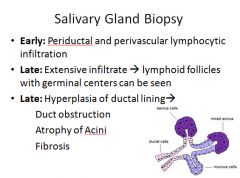
Sjogren Syndrome:
|
Characterized by dry eyes (keratoconjuctivitis sicca) and dry mouth (xerostomia), resulting from immune mediated destruction of the lacrimal and salivary glands.
It can occur as an isolated form (sicca syndrome) or more often associated with another autoimmune disease; RA, SLE, polymyositis, systemic sclerosis, vasculitis or thyroiditis. The ductal epithelial cells of the exocrine glands are the primary target. Most patients have autoantibodies to RNP proteins Ro (A) and La (B). Involved tissues show infiltration by lymphocytes and plasma cells. Most cases occur in women between 35-45 years of age. Salivary glands are often enlarged as a result of lymphocytic infiltrates. Extraglandular manifestations include synovitis, pulmonary fibrosis and peripheral neuropathy. *There is a 40-fold increased risk of developing a non-Hodgkin B cell lymphoma* |
|
|

|
|

|

|
|
|
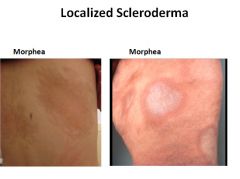
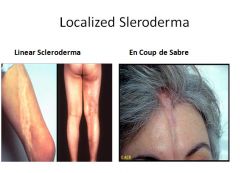
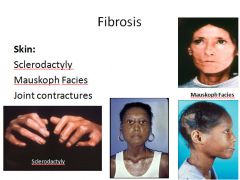
Systemic Sclerosis (Scleroderma):
|
Characterized by excessive fibrosis throughout the body and skin. Skin involvement is the usual presenting symptom, but it is the visceral involvement of the GI tract, lungs, kidneys, heart and muscles that produces the major morbidity and mortality.
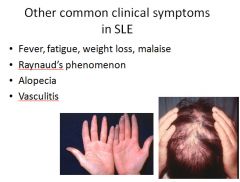
SS is classified into two groups: Diffuse - initial widespread skin involvement with rapid progression and early visceral involvement. Limited - Mild skin involvement of the fingers and face. Visceral involvement occurs late. Also called CREST syndrome; calcinosis, Raynaud phenomenon, esophogeal dysmotility, sclerodactyly and telangiectasia. |
|
|
Scleroderma etiology and pathogenesis:
|
Fibroblast activation with excessive fibrosis is the hallmark.
It is proposed that CD4+ T cells respond to an unidentified antigen and release cytokines that activate mast cells and macrophages leading to release of fibrogenic cytokines; IL-1, PDGF, TGF-b and fibroblast growth factors. Anti-Scl70 is found commonly in diffuse scleroderma. Anti-centromere is found commonly in limited scleroderma. Microvascular disease is also commonly present. Repeated cycles of endothelial damage followed by platelet aggregation lead to release of platelet PDGF that trigger periadventitial fibrosis and narrowing of the microvasculature with eventual ischemic injury. |
|
|
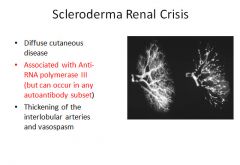
Scleroderma organ involvement:
|
Skin: Diffuse sclerotic atrophy of the skin, usually beginning in the fingers upper extremities.
GI: Fibrous replacement of the esophogeal muscularis leads to LES dysfunction, Barret's esophagus and strictures. May have loss of villi and microvilli in the intestines leading to malabsorption. MSK: Synovial hyperplasia and inflammation with later fibrosis. Lungs: May manifest as pulmonary hypertension (pulmonary vascular endothelial dysfunction) and or/interstitial fibrosis. Kidneys: Thickening of the interlobular arteries similar to the changes seen in malignant hypertension. Some patients do develop malignant hypertension as well; leads to fibrinoid necrosis. These patient die from renal failure. No specific glomerular changes. Heart: Patchy myocardial fibrosis (may cause arrythmia). Due to changes in the lungs, right ventricular hypertrophy and failure are common. |
|
|
Scleroderma clinical course:
|
SS affects more women than men at ages 50-60.
Almost all patients develop Raynauds. Progression collagenization of the skin leads to atrophy of the hands with increasing stiffness and complete immobilization of the joints. Difficulty swallowing, malabsorption, dyspnea and chronic cough (due to pulmonary changes), cor pulmonale, renal functional impairment and malignant hypertension are also seen. In most patients the disease is a slow steady downhill course over the span of many years. In patients without renal involvement, life span may be normal. The overall 10 year survival rate ranges between 35-70%. |
|

|
|
|
|
|
|

|
|
|

|

|
|

|

|
|

|

|
|

|

|
|
|
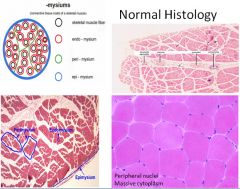
Inflammatory Myopathies clinical presentation:
|
Polymyositis and dermatomyositis;
Characterized by the gradual development of progressive motor weakness affecting the arms and legs, as well as the trunk, in assocation with histological evidence of muscle inflammation. While such inflammation predominantly involves striated muscle, smooth muscle and cardical muscle may be effected. The patient experiences increasing difficulty when rising from a chair, getting out of bed or climbing stairs. This may progress to difficulty reaching up or brushing the hair. At the most severe end of the spectrum, the patient may develop impairment of swallowing food and in full lung expansion. There is a predilection for interstital fibrosis and inflammatory arthritis to develop. |
|
|
Polymyositis/dermatomyositis etiology:
|
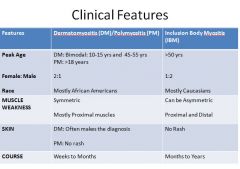
Both disorders are rare with women more effected than men.
Dermatomyositis has a bimodal age distribution; first peak occurs in childhood and second peak in mid and late adult life. Anti-Jo-1 = myositis Anti-Mi-2 = dermatomyositis |
|
|
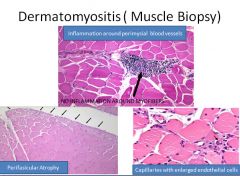
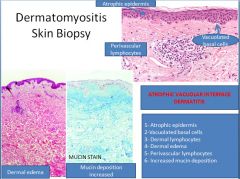
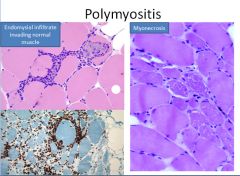
Polymyositis/dermatomyositis pathophysiology:
|
In polymyositis, inflammation is located around individual muscle fibers and the infiltrate is CD8+ T cell predominant.
In dermatomyositis, we see atrophy at the periphery of muscle bundles with a predominant CD4+ T cell infiltrate localized to the perifascicular space and surrounding capillaries (which are reduced in number). |
|
|
Polymyositis/Dermatomyositis clinical manifestations:
|
The hallmark symptom of both is weakness involving the upper and lower extremities.
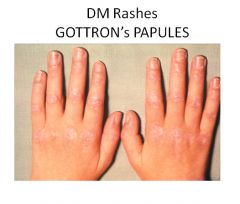
The four criteria for diagnosis of polymyositis are: 1) weakness 2) elevated CK 3) irritable elecromyogram 4) inflammatory infiltrate on histology For diagnosis of dermatomyositis a fifth criteria is: 5) skin rash (periorbital or in V-neck distribution on the trunk) Additional findings in dermatomyositis include red scaly eruptions of the MCP and PIP joints (Gottron's sign) and extensive sheets of muscle and soft tissue calcification. The diagnosis of an inflammatory myopathy usually means an increased risk of malignancy within 1-5 years. |
|
|
Polymyositis/Dermatomyositis treatment:
|
First line treatment is high dose corticosteroids.
Second line treatment is methotrexate, mycophenolate mofetile, IV Ig and rituximab |
|

|
|
|

|
|
|

|

|
|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|
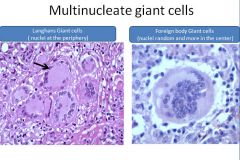
|
|
|
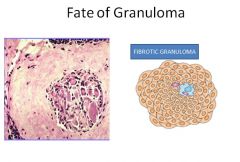
|
|
|

|
|
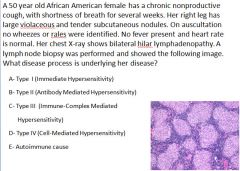
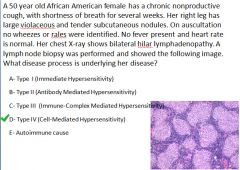
|
|
|
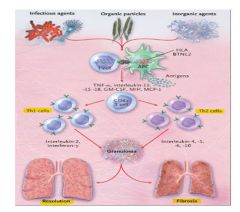
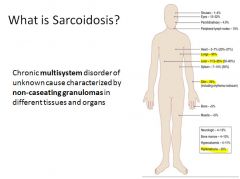
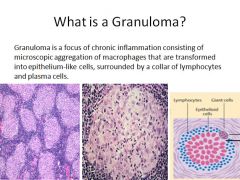
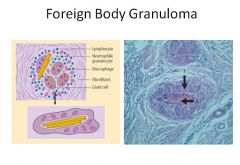
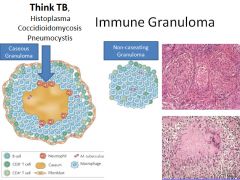
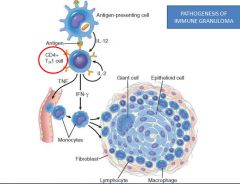
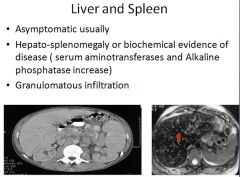
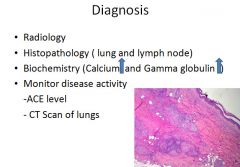
Pathogenesis of sarcoidosis
|

|
|
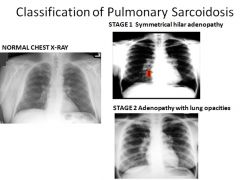
|
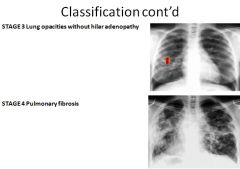
|
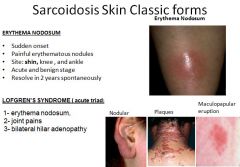
|
|
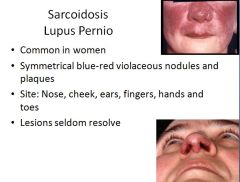
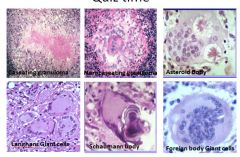
This lesion has nothing to do with actual lupus, just sarcoidosis.
|
|
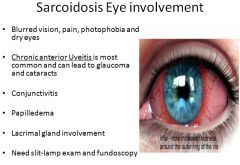
|
|
|
|

|
|

|

|
|

|

|
|

|

In addition to pain, most patients experience other symptoms, including fatigue, nonrestorative sleep, stiffness upon waking, and cognitive difficulties, specifically, impaired memory and concentration.
|
|
|
PET scan and MRI can show what two neurological abnormalities in patients with fibromyalgia?
|
Low serotonin levels and a reduction in thalamic metabolism.
|
|
|
Osteoarthritis overview:
|
The most common joint disorder. A frequent part of aging and cause of disability in persons over 65.
The fundamental feature of osteoarthritis is degeneration of the articular cartilage; any structural changes in the underlying bones are secondary. Usually appears insidiously. Knees and hands are common in women; hips in men. |
|
|
Osteoarthritis morphology:
|
The earliest changes include enlargement, proliferation and disorganization of chondrocytes. This process is accompanied by increasing water content of the matrix with decreasing concentration of proteoglycans.
Vertical and horizontal fibrillation and cracking of the matrix occur as the superficial layers of the cartilage are degraded. Eventually the bone is exposed. The bone becomes sclerotic and thickened. Cysts and mushroom shaped bony outgrowths develop on the exposed bone. |
|
|
Osteoarthritis pathogenesis:
|
The articular cartilage is maintained by chondrocytes producing proteoglycans and collagen type II. The cartilage undergoes constant degredation and replacement like adult bone. Any imbalance can lead to osteoarthritis.
Both mechanical stress and genetic factors can lead to development. Early in the disease, as cartilage degenerates, chondrocytes in deep layers proliferate and attempt to repair the damage. Although these changes are able to keep pace for a while, matrix changes and chondrocyte loss eventually predominate. |
|
|
Osteoarthritis clinical course:
|
An insidious disease affecting patients beginning in thier 50's and 60's.
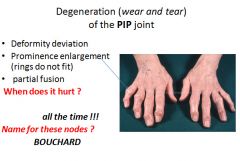
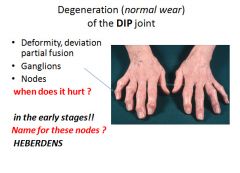
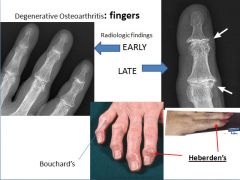
Characteristic symptoms include deep aching pain exacerbated by use, morning stiffness, crepitus, and limited range of movement. Osteophyte impingement on spinal foramina can cause nerve root compression with radicular pain, muscle spasms, muscle atrophy and neurologial deficits. Hips, knees, lower lumbar and cervical vertebrae, PIP and DIP joints of the fingers, and joints of the feet are commonly involved. Heberden nodes in the fingers, representing prominent osteophytes at the DIP, are characteristic in women. There is no way to halt the progression. With time joint deformity can occur, but unlike RA, fusion does not occur. |
|
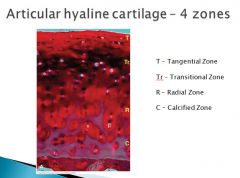
Tangential or gliding zone: This is the region closest to the articular surface, where chondrocytes are elongated, flattened and parallel to the long axis of the surface. Within this zone, a condensation of type II collagen fibers forms the so-called skin of the articular cartilage.
|
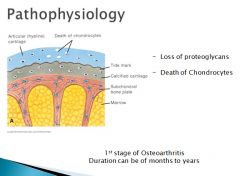
The death of chondrocytes lead to a crack in the articular cartilage that is followed by an influx of synovial fluid and further loss and degeneration of cartilage.
|
|
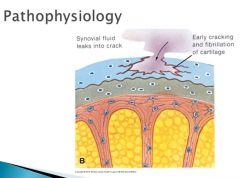
|
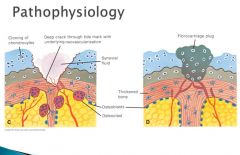
As a result of this process, cartilage is gradually worn away. Below the tidemark, new vessels grow in from the epiphysis, and fibrocartilage(D) is deposited
|
|
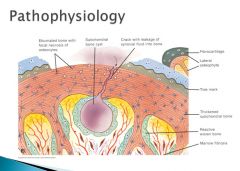
The fibrocartilage plug is not mechanically sufficient and may be worn away, thus exposing the subchondral bone plate, which becomes THICK and EBURNATED. IF there is a crack in this region, synovial fluids leak into the marrow space and produces a subchondral bone cyst. Focal regrowth of the articular surface leads to the formation of osteophytes.
|
|
|

|

|
|
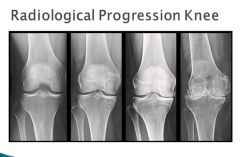
Osteoarthritis of the right knee. (a) normal. (b) Mild osteoarthritis with mild medial compartment osteophyte formation and loss of joint space. (c) Moderate osteoarthritis with osteophytic spurring, moderate joint space narrowing and subchondral sclerosis. (d) Severe osteoarthritis with subchondral sclerosis, severe joint space narrowing, large marginal osteophytes, and abnormal alignment of the knee.
|

|
|
|

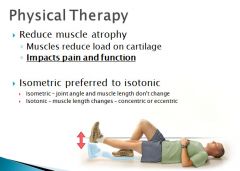
Besides NSAID's and Tramadol
|
|
|
|
|
|
|
|

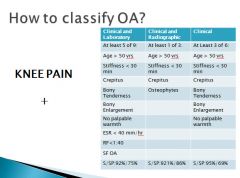
Of Osteoarthritis
|

Of Osteoarthritis
|
|
|
|
|
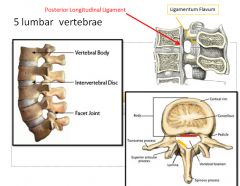
|
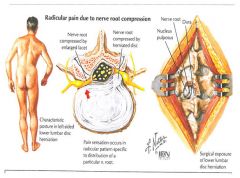
|
|
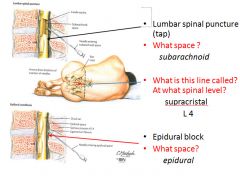
|
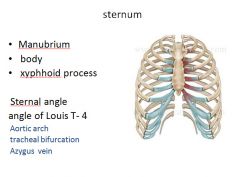
|
|
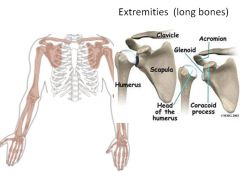
|
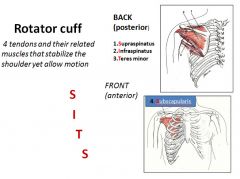
|
|
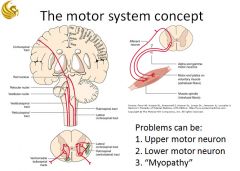
|

|
|
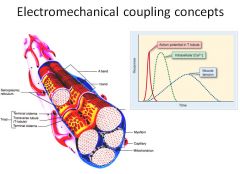
|
|
|
|

|
|

|

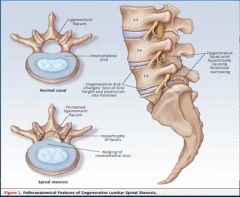
Chronic back pain is defined as pain present longer than 3 months.
|
|

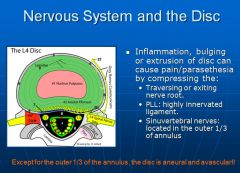
Radicular pain is due to irritation of the nerve roots—known. Axial pain can be due to a variety of sources-disc, facets, musculoskeletal, psychological, etc., and differentiated which of these is the problem is very difficult. As a result, treating axial pain is very unpredictable.
|
|
|
|
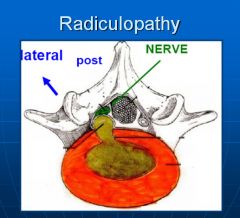
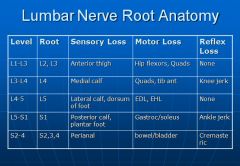
Radiculopathy is a result of irritation, usually mechanical pressure, on the nerve roots. It results in radiating pain down the buttocks and legs. The pattern of pain (dermatome) can be used to determine which level the impingement occurring.
|
|
|

|
|

|

|
|

|

|
|
|

|
|

|
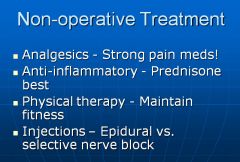
Can also treat with steroid injections for short term (2 months) relief, or do surgery to relieve pressure or fuse vertebrae.
|