![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
100 Cards in this Set
- Front
- Back
|
What is the difference between Type 1 & Type 2 respiratory failure? |
Type 1 --> hypoxic (low O2, normal or low CO2) Hypoxaemia + hyperventilation E.g. pulmonary oedema, pneumonia, asthma, pulmonary fibrosis, pulmonary thromboembolism Type 2 --> hypercapnic (low O2, high CO2) Implies a defect in ventilation with obstructed airways, decreased chest wall compliance, or CNS disease preventing excess CO2 from being exhaled. Restrictive disorder e.g.: - Lung disease e.g. fibrosis, collapse, oedema, consolidation - Pleural disease e.g. fibrosis, effusion, mesothelioma - Chest wall disease e.g. costospinal rigidity & deformity, abdominal splinting by obesity, ascites, or pregnancy - Neuromuscular disease e.g. muscular dystrophy, myasthenia gravis |
|
|
What are the clinical features of hypoxia? |
Dyspnoea, restlessness, agitation, confusion, central cyanosis If longstanding hypoxia --> polycythaemia, pulmonary hypertension, cor pulmonale |
|
|
What are the clinical features of hypercapnia? |
Headache, peripheral vasodilation, tachycardia, bounding pulse, tremor / flap, papilloedema, confusion, drowsiness, coma |
|
|
Describe the path of CN VII. |
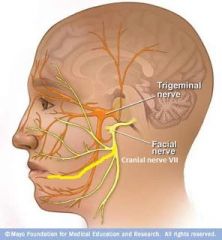
CN VII = facial nerve Arises from the cerebellopontine angle, lateral to the abducens nerve (CN VI - quite close to midline) & medial to the vestibulocochlear (CN VIII) nerve. Passes through the internal acoustic meatus & enters the facial canal (petrous part of the temporal bone). The facial canal has 3 segments: labyrinthine, tympanic, & mastoidal segments. CN VII leaves the facial canal via the stylomastoid foramen & lies immediately behind the parotid gland before dividing into 5 main branches (temporal, zygomatic, buccal, mandibular, cervical |
|
|
What oxygen saturation should you aim for in a patient with Type 2 respiratory failure? |
If evidence of CO2 retention on previous ABGs, give O2 cautiously, aiming for 88 - 92% O2 saturation. If no evidence of CO2 retention, aim for 93% O2 saturation. Never withhold oxygen from a hypoxaemic patient as the hypoxia is a bigger / more immediate problem than the potential hypercapnia. |
|
|
What are the three cardinal features of raised intracranial pressure? |
Headache, vomiting, & papilloedema. |
|
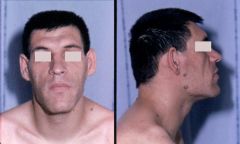
Which diagnostic facies is shown here? |
Acromegalic facies due to pituitary overproduction of growth hormone. Facial aspect of a patient with acromegaly. The nose is widened & thickened, the cheekbones are obvious, the forehead bulges, the lips are thick & the facial lines are marked. The forehead & overlying skin is thickened, sometimes leading to frontal bossing. |
|

Which diagnostic facies is shown here? |
Amiodarone facies. Note the deep blue discolouration around the malar area & nose. |
|

Which diagnostic facies is shown here? |
Cushingoid or moon facies. Other features of Cushing's Syndrome include abdominal obesity with thin limbs, purple or red striae, a round red face, buffalo hump, weak muscles, thin skin, hirsutism, irregular menstruation. |
|
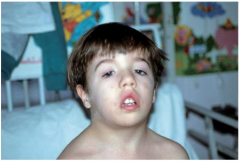
Which diagnostic facies is shown here? |
Myopathic facies. Note the elongated face, downslanting palpebral fissures, bilateral ptosis, facial weakness, open tent-shaped mouth, no/little facial expression, high arched palate, difficulty lifting the head when supine (due to weakness of the neck flexors), & sloping shoulders. Possible causes include myasthenia gravis, polymyositis, & many others. |
|

The facial rash shown is typical of which condition? |
Systemic lupus erythematosus. The rash is only seen in ~40% of SLE patients. A diagnosis of lupus requires 4 or more of the following: 1. Malar rash 2. Discoid rash 3. Photosensitivity 4. Oral ulcers 5. Non-erosive arthritis 6. Serositis (pleuritis or pericarditis) 7. Renal disorder (persistent proteinuria or cellular casts) 8. CNS disorder (seizures or psychosis) 9. Haematological disorder (haemolytic anaemia, leukopenia, lymphopenia, or thrombocytopenia) 10. Immunological disorder (Anti-dsDNA antibodies, Anti-Sm antibodies, or antiphospholipid antibodies) 11. ANA (+ve in >95% of cases) |
|
|
At what approximate level does bilirubin begin to be deposited in the tissues of the body, leading to discolouration (jaundice)? |
When the serum bilirubin level rise to about twice the upper limit of normal (normal total bilirubin: <20 umol/L |
|
|
What are some of the causes of cyanosis? |
Central cyanosis: 1. Decreased arterial oxygen saturation --> Decreased concentration of inspired oxygen (high altitude) --> Hypoventilation (coma, airway obstruction) --> Lung disease (COPD with cor pulmonale, massive PE) --> Right-to-left cardiac shunt (cyanotic congenital heart disease) 2. Polycythaemia 3. Haemoglobin abnormalities (rare) Peripheral cyanosis: 1. All causes of central cyanosis cause peripheral cyanosis 2. Exposure to cold 3. Reduced cardiac output (LV failure or shock) 4. Arterial or venous obstruction |
|
|
What are some of the causes of shock? |
1. Hypovolaemia --> External fluid loss (e.g. blood, vomit, diarrhoea, urine, burns, excessive sweating) --> Sequestration of body fluids in the abdomen (e.g. ascites), chest (e.g. haemothorax), or limbs (e.g. fracture) 2. Cardiac --> Pump failure (e.g. MI, acute mitral regurgitation) --> Cardiac tamponade --> Dissecting aortic aneurysm --> Arrhythmia 3. Massive PE 4. Sepsis (e.g. Gram -ve bacteria --> endotoxin) 5. Anaphylaxis 6. Endocrine failure (e.g. adrenal failure, hypothyroidism) 7. Neuropathic --> From drugs (e.g. antihypertensives, anaesthesia) --> Spinal cord injury --> Autonomic neuropathy |
|
|
Describe the different patterns of fever & give examples of possible aetiologies. |
1. Continued fever --> doesn't remit. Examples include typhoid fever, typhus, drug fever, malignant hyperthermia 2. Intermittent --> temperature falls to normal each day. Examples include pyogenic infections, lymphomas, miliary TB 3. Remittent --> daily fluctuations >2C, temperature doesn't return to normal. This is not characteristic of any particular disease 4. Relapsing --> temperature returns to normal for days before rising again. Examples include malaria (tertian 3-day pattern in which fever peaks every other day - P. vivax, P. ovale; quartan 4-day pattern in which fever peaks every 3rd day - P. malariae), lymphoma, Pel-Ebstein fever of Hodgkin's disease (very rare), pyogenic infection |
|
|
What is the normal body temperature range? |
36.5 - 37.2C Tympanic temperatures tend to be slightly lower than oral or rectal temperatures. |
|
|
When examining a patient with high blood pressure, is a Cushingoid appearance important? |
Yes. If the patient has hypercortisolism that is significant enough to cause hypertension, then they will almost always exhibit Cushingoid features, such as truncal obesity, facial fullness, dorsal hump etc. Although not specific (seen with significant weight gain from many causes), absence of these features probably excludes hypercortisolism. Patients with heavy alcohol intake, depression, & metabolic syndromes can assume similar appearances. |
|
|
When examining a patient with hypertension, is retinopathy an important feature? |
Yes. However, be aware that small artery disease & even retinal micro-haemorrhage / infarction are not specific to HTN (alse seen in diabetes, smokers, advanced age), severity grading is subjective & indirect ophthalmoscopy only provide limited views of the retina. Papilloedema, soft exudates, or flame haemorrhages in the abscence of diabetes indicate accelerated (malignant) HTN, which should be treated as a medical emergency. |
|
|
What are some of the non-drug / lifestyle treatments for hypertension? |
Weight reduction Reduced salt intake Increased fruit & veg intake Reduced alcohol intake Increased physical exercise Increased intake of omega-3 fatty acids |
|
|
In uncomplicated HTN, what are the first line antihypertensive agents? |
Begin with: - ACE-I or ARB - Dihydropyridine calcium channel blocker - Low dose thiazide diuretic (for patients >65yrs) Note: obese patients or those with features of metabolic syndrome will be more likely to develop diabetes on long term diuretics or beta blockers. |
|
|
What is a common side effect of ACE inhibitors? |
Cough Other side effects include angioedema, rash, gynaecomastia, neutropaenia, hepatitis, fatigue. |
|
|
How do ACE inhibitors work? |
ACE inhibitors work by reducing the levels of angiotensin II (vasoconstrictor), increasing the levels of vasodilator kinins, causing arteriolar (& to a lesser extent, venular) dilation, reducing aldosterone secretion which naturesis, & by a central effect that is not yet fully understood. |
|
|
How do ARBs work? |
Angiotensin receptor blockers reduce angiotensin-induced vasoconstriction, sodium reabsorption, & aldosterone release. |
|
|
Are beta blockers considered a first line treatment for uncomplicated HTN? |
No. They are associated with reduced protection against stroke & an increased risk of diabetes compared with other hypertensive classes. They are recommended for HTN when compelling indications, such as angina or post-MI, are present. |
|
|
What are the contraindications to beta blockers? |
Reversible airway disease e.g. asthma (can precipitate bronchospasm) Shock (cardiogenic or hypovolaemic) Bradycardia (HR < 45 - 50bpm) Second or third degree heart block Sick sinus syndrome (without pacemaker) Severe hypotension Uncontrolled heart failure |
|
|
How do beta blockers work? |
They lower BP by their negative inotropic effect on the left ventricle & by inhibition of beta-receptor mediated renin release. |
|
|
Which calcium channel blockers are non-dihydropyridines? |
Verapamil & diltiazem More cardiac effect than the dihydropyridine CCBs. |
|
|
Which calcium channel blockers are dihydropyridines? |
Nifedipine, felodipine, lercanidipine, & amlodipine. More peripherally acting than the non-dihydropyridine CCBs. |
|
|
How do calcium channel blockers work? |
They lower BP by: - Relaxing arterial & ateriolar wall smooth muscle - Inhibiting calcium ion influx (dihydropyridines more than verapamil & diltiazem) - Weakly inhibit cardiac myocytes (V&D more than dihydrop.) - Depress SA & AV nodes & conducting system (V&D only), which can lead to significant bradycardia |
|
|
What are the modifiable risk factors for cardiovascular disease? |
The modifiable risk factors contribute around 90% of the risk of MI observed worldwide are: - Blood lipid abnormalities - Smoking - Hypertension - Diabetes - Abdominal obesity - Psychosocial factors - Physical inactivity - Inadequate intake of fruits & vegetables |
|
|
With regards to CVD risk, above which weight circumference is the risk increased? |
>94cm for men >80cm for women |
|
|
What are the effects of bile acid binding resins? |
Useful for isolated hypercholesterolaemia (reduce LDL by 15 - 25%), may worsen hypertriglyceridaemia that is >3mmol/L. Indicated for hypercholesterolaemia, mixed hyperlipidaemia. Work by binding bile acids in the intestinal lumen, preventing reabsorption. Increased bile acid synthesis uses cholesterol, resulting in an increase in LDL uptake & removal from plasma. |
|
|
What are the effects of fibrates? |
Markedly reduce triglycerides (40 - 80%) & increase HDL (10 - 30%). In pure hypertriglyceridaemia, LDL may increase. Indicated for severe hypertriglyceridaemia with risk of pancreatitis, mixed hyperlipidaemia & dyslipidaemia associated with diabetes. |
|
|
What are the effects of statins? |
Most effective LDL-lowering agents (30 - 50% reduction). Reduces risk of MI, stroke, revascularisation procedures, & mortality in patients with high risk of CVD. Statins increase hepatic cholesterol uptake from blood, reduce concentrations of total cholesterol, LDL, & triglycerides (modest effect), & produce a small increase in HDL. Indicated for management of hypercholesterolaemia, mixed hyperlipidaemia, high risk of coronary heart disease, with or without hypercholesterolaemia. |
|
|
What are the potential side effects of statins? |
Common adverse effects are: - Myalgia - Mild transient GI symptoms - Headache - Insomnia - Dizziness - Elevated transaminases (dose dependent) Risk of myopathy & rhabdomyolysis are dose-related & also increased by illness & drug interactions. |
|
|
Which of the following statins has the least drug interactions?
A. Simvastatin B. Atorvastatin C. Rosuvastatin |
C. Rosuvastatin |
|
|
Which of the statins are most effective at reducing LDL levels? |
Rosuvastatin, simvastatin, & atorvastatin |
|
|
How often should lipid levels be checked when titrating the dosage of lipid-lowering medications? |
Every 4 - 6 weeks during dose titration then every 6 - 12 months during maintenance. |
|
|
What is the treatment for hypoglycaemia (BSL <4.0)? |
1. IV glucose (30ml of 50% dextrose, or 100ml of 20% glucose / 200ml of 10% glucose) over 15-30min 2. Glucagon 1mg IV/IM/SC 3. If awake, alert, and stable give hypo pack (quick acting carbohydrates) followed by some complex long acting carbs |
|
|
What are some common causes of hypoglycaemia?
|
Reduced oral intake with usual diabetic meds Excess diabetic therapy / insulin Sepsis Liver failure Alcohol excess (will need IV thiamine prior to IV glucose to prevent precipitating Wernicke encephalopathy - remote risk) Fasting |
|
|
What is the dose of naloxone in suspected opiate overdose?
|
400 - 800 mcg IV/IM/SC repeated as necessary until respiratory rate improves. Reconsider diagnosis if there is no response after a total of 10 mg has been given. Watch for recurrence of sedation when naloxone wears off (half life of naloxone is <1hr) |
|
|
What is the treatment of benzodiazepine overdose?
|
Flumazenil 200mcg IV over 15sec If needed, further doses of 100mcg can be given at 1min intervals up to a total dose of 2mg. Note that the half life of flumazenil is ~45-60min, after which sedation may re-ocur. Flumazenil is rarely needed as benzodiazepine toxicity is usually mild and flumazenil can precipitate seizures and acute benzo withdrawal (particularly in those with a history of epilepsy, co-ingestion of pro-convulsant drugs such as TCAs, or benzodiazepine dependent patients). Flumazenil is indicated when needed to avoid intubation (especially in kids, the elderly, and those with respiratory diseases; to reverse procedural sedation; if I+V are not available). Note that hypotension is uncommon after benzodiazepine poisoning. If present, it suggests either co-ingestion of another drug or an underlying cardiac pathology. |
|
|
What is LDH? |
Lactate dehydrogenase is an important enzyme of the anaerobic metabolic pathway. |
|
|
What causes elevated LDH in the blood? |
LDH is a non-specific marker of tissue turnover and also released when there is tissue damage with loss of cytoplasm. It is present in high concentrations in muscle, liver, and kidney. It is present in moderate concentrations in red blood cells. Conditions that can cause elevated LDH include: - Haemolysis of red blood cells - Hepatitis - Acute renal failure - Anaemia - Pulmonary embolism - Myocardial infarction - Pancreatitis - Intracranial haemorrhage - Bone fractures - Muscle trauma - Cancers - Cellular necrosis - Infections such as encephalitis, meningitis, encephalitis, and HIV.
|
|
|
What are some non-blood body fluids that can be tested for LDH and why? |
1. LDH increases during effusion in serous body fluids such as pericardial, pleural, and peritoneal fluids, and can serve to characterise the effusion (e.g. Light's criteria to differentiate between transudative and exudative pleural effusion) 2. LDH levels in CSF can help differentiate between bacterial (elevated LDH) and viral (normal LDH) meningitis |
|
|
What is Light's criteria? |
Light's criteria allow for the differentiation of transudative vs exudative pleural effusion. It is more accurate for the diagnosis of exudative effusions. The fluid is considered an exudate if any of the following are present: 1. The ratio of pleural fluid to serum protein is >0.5 2. The ratio of pleural fluid to serum LDH is >0.6 3. The pleural fluid LDH value is greater than two-thirds the upper limit of the normal serum value Another diagnostic criteria for pleural effusion in patients with normal serum protein is simply: Transudate = protein <30g/L Exudate = protein >30g/L |
|
|
What is oxidative stress? |
Oxidative stress refers to cellular abnormalities that are induced by reactive oxygen species (ROS), which belong to a group of molecules called free radicals. |
|
|
What are free radicals? |
Free radicals are highly reactive molecules with an unpaired electron in an outer orbit. They react to with all inorganic and organic molecules to remove electrons and convert them into free radicals. The biologically important free radicals are ROS (reactive oxygen species) and nitric oxide. The generation of free radicals is increased by exposure to UV light, radiation and toxins, during normal cellular aging, and oxygen deprivation. |
|
|
What are reactive oxygen species (ROS)? |
ROS is a free radical, they are produced normally in small amounts in all cells during the reduction-oxidation (redox) reactions that occur during cellular respiration and energy generation (superoxide --> hydrogen peroxide). They are produced in phagocytic leucocytes (mainly neutrophils and macrophages) to destroy ingested microbes etc during inflammation. The enzymes glutathione peroxidase and catalase breaks down the hydrogen peroxide. Increased amounts of free radicals during pathologic injury overwhelms these scavenging systems. |
|
|
What are the functional and morphogical consequences of hypoxia and ischaemia on the cells? |
Oxygen deficiency leads to reduced generation of ATP and failure of energy-dependent cellular systems. Oxygen deficiency --> decreased oxidative phosphorylation --> decreased ATP, which leads to: 1. Reduced activity of ATP-dependent cell membrane Na/K pump --> influx of Ca, H2O, and Na + efflux of K --> swelling of ER, cellular swelling, loss of microvilli, blebs. 2. Increased anaerobic glycolysis --> decreased glycogen, increased lactic acid, and decreased pH which reduces activity of many intracellular enzymes. 3. Detachment of ribosomes leading to decreased protein synthesis Hypoxia may increase production of ROS, which have many damaging effects. If lysosomal and mitochrondrial membranes are damanged, lysosomal acid hydrolases are activated by low pH and the cell begins to digest itself (necrosis). |
|
|
What is ischaemia-reperfusion injury? |
Restoration of blood flow to ischaemic tissue may exacerbate the tissue injury. This is likely due to increased production of ROS by damaged cells and leucocytes that have been recruited to mop up necrotic cells. These inflammatory cells + complement proteins may contribute to tissue injury through inflammatory reactions. |
|
|
What are some diseases that are caused by misfolded proteins? |
Misfolded proteins can accumulate in the endoplasmic reticulum and stress the adaptive mechanisms, resulting in apoptosis. Diseases caused by misfolded proteins that are degraded, leading to their deficiency: - Cystic fibrosis - Familial hypercholesterolaemia - Tay-Sachs Disease Diseases caused by misfolded proteins that cause ER stress-induced cell loss: - Retinitis pigmentosa - Creutzfeld-Jakob Disease - Alzheimers Diseases caused by misfolded proteins due to both ER stress-induced cell loss + functional deficiency of the protein: - Alpha-1-antitrypsin deficiency |
|
|
What are some of the physiological and pathological cellular adaptations to stress? |
Hypertrophy (increase in size of cells) Hyperplasia (increase in number of cells) Atrophy (decrease in number of cells) Metaplasia (change in cell type) |
|
|
What are the main pathways for pathological accumulations in cells? |
Cells may accumulate abnormal amounts of various substances, which may be benign or may cause varying degrees of injury. The main pathways are: 1. Inadequate removal and degradation of an endogenous substance 2. Excessive production of an endogenous substance 3. Deposition of an abnormal exogenous substance material |
|
|
What are the four main structural components of the kidney?
|
Glomeruli --> functional units that filter blood Tubules --> control the amount of fluid, ions, and molecules excreted / reabsorbed by the kidneys Interstitium --> the supporting scaffold Blood vessels --> arterial and venous circulation |
|
|
What are the 5 major glomerular syndromes?
|
1. Nephritic Syndrome --> haematuria, azotaemia, variable proteinuria, oliguria, oedema, and HTN 2. Rapidly progressive glomerulonephritis --> acute nephritis, proteinuria, acute renal failure 3. Nephrotic Syndrome --> proteinuria (>3.5g/day), hypoalbuminaemia, hyperlipidaemia, lipiduria 4. Chronic renal failure --> azotaemia progressing to uraemia over months to years 5. Isolated Urinary Abnormalities --> glomerular haematuria &/or subnephrotic proteinuria |
|
|
What is azotaemia and what is the difference between azotaemia and uraemia? |
Azotaemia is an increase in urea / BUN with an associated increase in creatinine.
- Note: in America, only the nitrogen component of urea is assayed, giving a blood urea nitrogen (BUN) level. In Europe the whole urea molecule is assayed. Urea is ~2x the level of BUN. Uraemia is the term used when azotaemia is severe enough to cause clinical signs and symptoms. |
|
|
What is that most common mechanism of acute kidney injury? |
AKI is most often caused by severe tubular injury (previously called acute tubular necrosis), but may also result from acute, usually severe, injury to glomeruli, vessels, or interstitium. |
|
|
What is the pathophysiology of nephrotic syndrome? |
Derangement in the capillary walls of the glomeruli that results in increased permeability to plasma proteins and proteinuria (urinary protein loss >3.5g/24hrs). Protein loss --> hypoalbuminaemia --> reduced plasma colloid osmotic pressure --> transudate of fluid across small blood vessels --> generalised oedema. Hyperlipidaemia is also frequently seen. |
|
|
Briefly describe Nephritic Syndrome. |
Nephritic Syndrome is caused by inflammatory lesions of the glomerulus characterised by haematuria, azotaemia, and hypertension. It is seen in diseases in which glomerular inflammation is prominent. Proteinuria may be modest or completely absent. |
|
|
What are the common mechanisms of glomerular injury? |
Most glomerular diseases are immunological in origin, due to deposition of immune complexes or antibodies in the glomerular capillary wall. 1. Immune complex deposition --> either deposition of Ag-Ab complexes that were formed in circulation (e.g. in SLE) or Abs reacting in situ within the glomerulus (either with fixed glomerular antigens or extrinsic molecules that have wound up in the glomerulus). May result in local inflammation (complement system activation with leucocyte recruitment) - glomerulonephritis with nephritic syndrome, or disruption of glomerular permeability barrier without inflammation (nephrotic syndrome). 2. Deposition of anti-GBM antibody --> antibodies that bind to antigens in the glomerular basement membrane. The subsequent injury is caused by inflammation triggered by the complement system and Fc-receptor dependent mechanisms, as in immune complex mediated disease 3. Other mechanisms of glomerular injury --> defective regulation of complement activation, injury to podocytes by toxins etc, abnormal glomerular permeability (e.g. due to genetic mutations). |
|
|
What is Minimal Change Disease?
|
Minimal change disease is the most common cause of nephrotic syndrome in children; its unique feature is the absence of glomerular pathology by light microscopic evaluation. |
|
|
What is the most common cause of nephrotic syndrome in children? |
Minimal change disease. |
|
|
What is the pathogenesis of minimal change disease?
|
Unknown - no deposits of antibodies or immune complexes are seen in the glomerulus, and no sign of inflammation. It is suspected that a permeability-inducing circulating factor causes the leakiness of the GBM (glomerular basement membrane) to albumin.
|
|
|
What is the morphology of minimal change disease under microscopy? |
The glomeruli appear normal by light microscopy, however electron microscopy shows diffuse effacement of podocyte foot processes (unclear if this is the cause or consequence of the proteinuria). Fluorescent microscopy is negative. |
|
|
What are the clinical features of minimal change disease? |
Typically presents with nephrotic syndrome in a previously healthy child, typically between 1 and 7 years old. Classically the protein loss is selective, with only low molecular weight proteins such as albumin being lost. Majority of patients respond well to corticosteroid therapy, but proteinuria recurs in 60% of initial responders, many of whom become steroid dependent. In adults the response to steroids is slower and relapses are more common. |
|
|
What is membranous nephropathy?
|
Membranous nephropathy is a renal disease caused by immune complex deposition in the glomerular capillary wall, resulting in nephrotic syndrome. |
|
|
What is the pathogenesis of membranous nephropathy? |
In most cases the immune complexes are formed in situ, with auto-antibodies binding to endogenous podocyte antigens (e.g. phospholipase A2 receptor) or planted antigens. The aetiology of this autoimmune condition is unknown. In up to 80% of cases, membranous nephropathy is the primary condition, with no associated disease. Less commonly it may be seen in association with other autoimmune diseases such as SLE. May also be secondary to infections (e.g. malaria, syphilis, Hep B) or tumours (e.g. melanoma, lung Ca, colon Ca). May occur secondary to some medications (e.g. penicillamine, captopril, NSAIDs). All of these conditions may cause formation of antibodies that bind to antigens planted in the GBM. The formation of subepithelial immune deposits leads to complement activation on the surface of the podocytes and generates the membrane attack complex (C5 - C9), which in turn causes podocyte and GBM injury and proteinuria. |
|
|
What is the morphology of membranous nephropathy on histology? |
Light microscopy: the main finding is diffuse thickening of the GBM, caused by deposition of immune complexes Fluorescence microscopy: granular deposits of IgG and C3, diffuse. Electron microscopy: subepithelial deposits with interspersed spikes of GBM material. Podocyte foot processes are diffusely effaced, as in other diseases with proteinuria. There is typically no inflammation. |
|
|
What are the clinical features of membranous nephropathy? |
Membranous nephropathy usually presents in adults between 30 and 60 years old, and follows an indolent and progressive course. Onset is sudden, characterised by nephrotic syndrome, usually without any antecedent illness. Proteinuria is non-selective, leaking both large and small proteins into the urine. Does not respond well to corticosteroids, usually immunosuppressive drugs such as cyclophosphamide and cyclosporin are used. Patients who go into spontaneous remission or remission following treatment tend to do well, although ~20% have a remitting and relapsing course, with 10% going on to develop renal failure. |
|
|
What is the difference between granular and linear immunofluoresence (with regards to glomerular injury)? |
Granular immunofluorescence: due to deposition of circulating immune complexes onto subepithelial, mesangial, or subendothelial surfaces. May also occur when antibodies bind to insitu antigens on podocytes causing subepithelial immune complex formation (membranous glomerulonephritis). Linear immunofluorescence: occurs due to antibodies against the glomerular basement membrane (GBM), causing anti-GBM glomerulonephritis (no immune complexes formed) |
|
|
What is focal segmental glomerulosclerosis? |
Focal segmental glomerulosclerosis (FSGS) is a common cause of nephrotic syndrome, it is characterised by sclerosis of a portions of glomeruli. May occur as primary disease or secondary to other diseases. |
|
|
What is the pathogenesis of focal segmental glomerulosclerosis (FSGS)? |
Disease is thought to be initiated by injury to podocytes, although the cause of that initial injury remains unknown. Possibly due to circulating factors that have not yet been indentified. Most of the time FSGS is primary, but may develop secondary to HIV, heroin abuse, other forms of glomerular disease, or inherited defects if cytoskeletal or podocyte proteins. May also be seen where there is reduced renal mass (e.g. due to ablation or disease) due to increased blood flow to the remaining kidney causing haemodynamic injury to the glomeruli. |
|
|
What is the morphology of glomerular pathology in focal segmental glomerulosclerosis (FSGS)?
|
Light microscopy: focal and segmental sclerosis and accumulation of matrix material ('hyaline') in the mesangium, that obliterates the glomerular capillaries, and throughout the abnormal segment. Fluorescence microscopy: focal IgM + C3 in some cases, reflecting non-specific trapping (no immune complexes) Electron microscopy: effacement of podocyte foot processes As the name suggests, not all glomeruli are affected, and those that are only have portions of sclerosis - it does not affect the entire structure |
|
|
What are the clinical features of focal segmental glomerulosclerosis (FSGS)? |
The classic presentation of FSGS is nephrotic syndrome, sometimes associated with microscopic haematuria and hypertension. The proteinuria is non selective (both small and large size proteins are lost in urine), and the response to immunosuppressive meds is poor: ~50% of patients develop ESRF within 10 years.
|
|
|
What is the P/F ratio? |
The P/F ratio is a tool to identify acute hypoxemic respiratory failure when supplemental oxygen has already been administered and no room air ABG is available, or pulse oximetry readings are unreliable. The diagnostic criteria for acute hypoxemic respiratory failure is: - PaO2 < 60 mmHg on room air measured by ABG, or - SpO2 < 91% on room air measured by pulse oximetry, or - P/F ratio < 300 on oxygen The P/F ratio indicates what the PaO2 would be on room air (if patient was taken off oxygen): P = PaO2 from the ABG F = FiO2 expressed as a decimal e.g. PaO2 = 90 on 40% oxygen (FIO2 = 0.40): 90 / 0.40 = P/F ratio = 225. A normal P/F Ratio is ≥ 400 and equivalent to a PaO2 ≥ 80 mmHg. |
|
|
What is calciphylaxis? |
Calciphylaxis is a rare and serious disorder that presents with skin ischemia and necrosis and is characterized histologically by calcification of arterioles and capillaries in the dermis and subcutaneous adipose tissue. It is a lethal disease that carries a high morbidity and mortality, with an estimated six-month survival of approximately 50 percent. There is no approved treatment for calciphylaxis. Calciphylaxis most commonly occurs in patients who have end-stage kidney disease (ESKD) and are on dialysis but may also occur in kidney transplant recipients and in non-ESKD patients. |
|
|
What is Hungry Bone Syndrome?
|
Hungry Bone Syndrome is a condition that can occur in ESKD patients post parathyroidectomy. While hypocalcaemia post parathyroidectomy is usually transient, in these patients it is severe and prolonged, despite normal or elevated levels of PTH. In addition to hypocalcaemia, you may also see hypophosphataemia, hypomagnesaemia, and hyperkalaemia - which likely reflects increased bone influx and efflux. |
|
|
What does parathyroid hormone do? |
Parathyroid hormone is involved in calcium and phosphate homeostasis. PTH is secreted in response to hypocalcaemia, it facilitates the synthesis of active vitamin D (calcitriol: 1,25-dihydroxycholecalciferol) in the kidneys. PTH effect on bones: stimulates bone resorption by osteoclasts to release calcium into the blood (also stimulates osteoblasts to differentiate into osteoclasts, and prevents osteoblast formation). PTH effect on kidneys: PTH acts on the kidneys to increase serum calcium in three different ways. It acts on the distal convoluted tubule and collecting duct, directly increasing calcium reabsorption. PTH also decreases phosphate absorption at the proximal convoluted tubule (phosphate ions in serum react with calcium to form salts that are insoluble, resulting in decreased serum calcium, so reducing phosphate ions allows for more ionised calcium in the blood). PTH effect on small intestines and reabsorption of calcium: PTH indirectly promotes reabsorption of calcium by the small intestines. This is due to PTH stimulating production of 1alpha-hydroxylase at the proximal convoluted tubule, which catalyses the synthesis of active Vit D (1,25-dihydroxycholecalciferol). Active Vit D helps with calcium reabsorption in the DCT and the small intestines). |
|
|
What is the serum half life of activated parathyroid hormone?
|
The serum half-life of activated PTH is only a few minutes - it is removed quickly from the serum by the kidneys and liver. |
|
|
What are the different types of hyperparathyroidism? |
Primary --> abnormality of the parathyroid gland itself, causing over secretion of PTH. Secondary --> refers to the compensatory oversecretion of PTH in response to abnormally low calcium due to other pathologies (e.g. renal failure, GI malabsorption, or Vit D deficiency). Tertiary --> exceedingly rare, it is seen in the context of continuous PTH secretion even after the condition causing secondary hyperparathyroidism has resolved |
|
|
What are the features of primary hyperparathyroidism? |
Labs: high PTH, hypercalcaemia, hypophosphataemia Symptoms: excessive thirst and urination, constipation, bone pain, fatigue, depression, and possible kidney stones (stones, bones, groans, thrones, and psychiatric overtones) Primary hyperparathyroidism is most often due to adenoma, hyperplasia, or more rarely a carcinoma. |
|
|
What are the features of secondary hyperparathyroidism?
|
Depends on the underlying process. Secondary hyperparathyroidism is due to a compensatory oversecretion of PTH in response to abnormally low calcium due to other pathologies such as renal disease, GI malabsorption, or Vit D deficiency.
In chronic renal failure there will be high PTH, low calcium, and high phosphate. In GI malabsorption or Vit D deficiency there will be high PTH with low calcium and phosphate. |
|
|
What are the features or tertiary hyperparathyroidism?
|
Tertiary hyperparathyroidism is exceedingly rare but is seen in the context of continuous PTH secretion even after a secondary hyperparathyroidism precipitating condition is resolved. There will be moderately high PTH, normal or high calcium, and low phosphate |
|
|
What are the features of hypoparathyroidism?
|
Clinical features due to hypocalcaemia, such as abdominal pains, muscle cramping, and paraesthesias. May be positive for Chvostek and Trousseau signs. Chvostek sign --> positive when the cheek is tapped lightly and the face contracts on the same side (facial nerve is hyperexcitable due to hypocalcaemia). Trousseau sign --> positive when a BP cuff is inflated to >SBP and left on for 3min, causing muscle spasms of hand and forearm (brachial artery occlusion allows the hypocalcaemia to induce nerve excitability). Labs: low PTH, low calcium |
|
|
What is the role of calcium in the body?
|
Calcium is a divalent cation essential to heart, kidney, bone, and nervous system functioning. Ca has a vital role in cardiac contractions - excess intracellular Ca results in increased contractility, low intracellular Ca results in decreased contractility which can lead to prolonged QT intervals. Extreme hypercalcaemia can lead to very short QT intervals. This can precipitate possibly fatal arrhythmias such as VT/VF. Hypocalcaemia causes nerve hyperexcitability at every level of neurons throughout the nervous system (decreases the threshold potential). Hypercalcaemia can precipitate renal stones. |
|
|
What are the key priorities in the management of decompensated critically unwell chronic liver disease patients? |
1. Recognise and treat sepsis, especially spontaneous bacterial peritonitis 2. Prevent and treat hepatic encephalopathy 3. Prompt management of bleeding 4. Volume resuscitation 5. Management of electrolytes 6. Identification of alcohol dependence and withdrawal 7. VTE prophylaxis --> based on INR and platelets have a coagulopathy but these pts are actually prothrombotic |
|
|
What is the mainstay of treatment for variceal bleeding in ICU admissions? |
ICU admissions relating to bleeding tend to have a good survival rate even in the setting of organ failures. Mainstay of treatment: - Vasoactives (octreotide, terlipressin) - Abx - Early endoscopy (<12hrs) NOTE: do NOT give bleeding cirrhotic patients tranexamic acid (no benefit in setting of GI bleeding, even if variceal, and is associated with harms such as portal vein thrombosis and DVT). Also should not give fresh frozen plasma as cirrhotics have balanced coagulation defects (lose both pro- and anticoagulant factors) and variceal bleedingis precipitated by portal pressure, not coagulopathy. FFP associated with worse outcomes. |
|
|
What is hepatorenal syndrome?
|
Hepatorenal syndrome is a diagnosis of exclusion and represents the end-stage of a sequence of reductions in kidney perfusion induced by increasingly severe hepatic injury. Note that hepatorenal syndrome is one of many possible causes for AKI in patients with acute or chronic liver disease. |
|
|
Should all patients with ascites have a diagnostic tap? |
10% of patients who present to hospital with ascites have spontaneous bacterial peritonitis. Diagnostic taps are quite safe even in coagulopathy, and missing an SBP translates to 65% in-hospital mortality rate. |
|
|
What is the management of hepatic encephalopathy?
|
... |
|
|
What are the features of hepatorenal syndrome? |
Patients typically present with oedema, oliguria / anuria, and ascites in the setting of acute or chronic liver disease. They will also have low urinary sodium (<10 mEq/L). Diagnostic Criteria (need all 6): Hepatic failure (cirrhosis + ascites) Renal failure - AKI (creat ?15mg/dl) Absence of other causes of renal failure: - Absence of hypovolaemia (nil improvement in creatinine post fluid challenge or withdrawal of nephrotoxics) - Absence of intrinsic renal disease (normal radiology, nil or low haematuria / proteinuria - Absence of sepsis - Absence of nephrotoxics |
|
|
What is the patholophysiology of hepatorenal syndrome?
|
Cirrhosis and portal hypertension --> splanchnic vasodilation --> effective arterial hypovolaemia (not a true hypovolaemia) --> decreased renal blood flow --> activation of renin-angiotensin-aldosterone system --> increased renal absorption of Na and water ( leads to ascites) + renal vasoconstriction (leads to hepatorenal syndrome) |
|
|
What are some causes of secondary osteoporosis? |
Endocrine causes: - Cushing's syndrome, hypogonadism, thyrotoxicosis, hyperparathyroidism Drug causes: - Glucocorticoids, heparin, anticonvulsants (e.g. phenytoin), immunosuppressants, thiazolidinediones, oncology (e.g. aromatase inhibitors, androgen deprivation therapy) Chronic diseases: - Renal impairment, liver cirrhosis, malabsorption, chronic inflammatory polyarthropathies (e.g. RA) Other causes: - Nutritional deficiency (e.g. anorexia), MM and malignancy, osteogenesis imperfecta, post-gastrectomy / gastric bypass surgeries |
|
|
What are the risk factors for osteoporosis? |
Major risk factors: - Hx of minimal trauma fracture - Female - >70 years old - Hx of falls - Parental Hx of hip fracture - Premature menopause or hypogonadism - Prolonged use of glucocorticoids - Use of other meds that cause bone loss - Low body weight - Low muscle mass and strength - Low physical activity - Prolonged immobility - Poor balance Other risk factors: - Smoking - High EtOH intake - Energy, protein, or Ca undernutrition - Vitamin D insufficiency / deficiency |
|
|
What are some of the medications that can cause bone loss? |
Glucocorticoids, excessive thyroid hormone replacement, anti-androgen therapy, anti-oestrogen treatments (aromatase inhibitors), SSRI, thiazolidenediones, PPI, certain anti-epileptic drugs, and certain anti-psychotics |
|
|
When does most bone loss occur in glucocorticoid induced osteoporosis? |
Bone loss occurs most rapidly in the first 6 - 12 months of oral glucocorticoid therapy Prednisolone >5mg daily (or equivalent) for more than 3 months is associated with osteoporosis. These patients should be screened and osteoporosis therapy started if they have risk factors. |
|
|
In which situations would a BMD assessment be appropriate? |
Younger than 50: - Minimal trauma # (case by case basis) - Disease or condition assoc w/ bone loss 50 - 60 years old: - Vertebral # (not assoc w/ trauma) - Peripheral minimal trauma # (case by case basis) - Disease or condition assoc w/ bone loss - Meds increasing bone loss 60 - 70 years old: - As above plus the following - Hip # in a parent - Underweight - Multiple falls - Immobility |
|
|
What tools can you use to calculate individual fracture risk in patients with osteoporosis? |
Garvan Fracture Risk Calculator FRAX (Fracture Risk Assessment Tool) |
|
|
What are bone turnover markers and when should they be used? |
Markers of resorption include serum C-telopeptide (CTX), urinary deoxypyridonoline (DPD), urinary N-telopeptide (NTX)/ Markers of formation include P1NP (N-terminal propeptide of type 1 procollagen), bone specific alkaline phosphatase, and osteocalcin. CTX and P1NP are most often used. They should be used to monitor effect of osteoporosis Rx, but should not be used for diagnosis. |