Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
41 Cards in this Set
- Front
- Back
|
what is the role of CD71 in erythroid precursor cells in the marrow
|
CD71 is the transferrin receptor. it allows the erythroid precursor to receive iron from the recycling pool
|
|
|
ascorbic acid facilitates iron absorption, while tannins inhibits it
|
true
|
|
|
24of with iron deficiency anemia (Hb6.3g/dL, Fe 12micrograms/dL). you advise her that the best test for monitoring a response is HEMOGLOBIN. the earliest she might check for a response is after the FIFTH DAY. after the first week the Hb can be expected to rise by 0.1 TO 0.2 G/dL per day for the next 3 weeks
|
24of with iron deficiency anemia (Hb6.3g/dL, Fe 12micrograms/dL). you advise her that the best test for monitoring a response is HEMOGLOBIN. the earliest she might check for a response is after the FIFTH DAY. after the first week the Hb can be expected to rise by 0.1 TO 0.2 G/dL per day for the next 3 weeks
|
|
|
how is zinc protoporphyrin (ZPP) level used to distinguish between iron deficiency and thalassemia
|
ZPP is elevated (>99micrograms/dL) in iron deficiency and normal (10-99 micrograms/dL) in thalassemia
|
|
|
what changes are found in marrow in megaloblastic anemia?
marrow cellularity: number or erythroid precursors: myeloid : erythroid ratio: |
marrow cellularity: hyperplastic
number or erythroid precursors: increased myeloid :erythroid ratio: decreased |
|
|
what molecule serves as the cheif transport protein for cobalamin and is lacking in severe megaloblastic anemia of infancy?
|
transcobalamin II (TCII)
**transcobalamin is an acute phase reactant |
|
|
describe the two types of antibodies to the intrinsic factor (IF) seen in pernicious anemia
|
the blocking antibodies block the binding of cobalamin to intrinsic factor in the stomach
the binding antibodies bind to the cobalamin-IF complex, preventing it from binding to the ileal receptors in the small bowel |
|
|
39 yom immigrant from Finland* has recently been diagnosis with megaloblastic anemia. Serologic tests for autoAb are negative. what parasite can cause his anemia?
|
Diphyllabothrium latum
serum cobalamin will be decreased, urine methylmalonic acid will be increased |
|
|
27 yom with celiac disease. the patient is receiving monthly injections of vit B12 and oral folate.
his serum B12 is 327pg/ml (160-950) and serum folate is 5.1ng/ml (>3.5). is red cell folate indicated? |
no. vit B12 is required for folate to gain entry into the cells, but both B12 and serum folate are within normal range, so it is unlikely that red cell folate would be abnormal; vitamin replenishment is sucessful in thei patient
|
|
|
what is a likely red cell and iron studies profile in a 62yof with an acute flare of her rheumatoid arthritis?
MCV: MCH: RDW: Serum Fe: TIBC: %iron sat: Ferritin: free erythrocyte porphyrin: |
MCV: nml
MCH: nml RDW: nml Serum Fe: low TIBC:nml or low %iron sat: low Ferritin: high free erythrocyte porphyrin:nml |
|
|
some patients taking chloramphenicol develop reticuloctopenia anemia, neutropenia and thrombocytopenia as well as morphological changes in the marrow. what is the outcome for most patients?
|
transient pancytopenia occurs in about half of the patients, but only a small percentage go on to develop irreversible pancytopenia
|
|
|
does serology identify the specific hepatitis virus for most cases of aplastic anemia? what are the demographics for the typical patient?
|
no, most are seronegative for hep A, B, C and G. the patients are usually male under 20 years of age
|
|
|
thymomas are associated with with both pure red cell aplasia and aplastic anemia
|
thymomas are associated with with both pure red cell aplasia and aplastic anemia
|
|
|
both alkylating agents and DNA crosslinking agents readily cause breaks in the DNA of Franconi's patients
|
both alkylating agents and DNA crosslinking agents readily cause breaks in the DNA of Franconi's patients
|
|
|
both Franconi's and Diamond -Blackfan patients have increased HbF
|
both Franconi's and Diamond -Blackfan patients have increased HbF
|
|
|
What is the most common cause of reversible acquired sideroblastic anemia?
other causes although less common? |
ethanol
antiTB drugs--isoniazid, cycloserine and pyrazinamide lead is the key toxin |
|
|
what is the bacterial agent responsible for hemolytic anemia Oroya fever?
|
Bartonella bacilliformis
|
|
|
what two tests are used to diagnose congential dyseythopoieetic anemia type II(CDA-II)? give the results of each
|
acidified serum hemolysis test is positive
the sucrose hemolysis test is negative CDA-II red cells react strongly with anti-I and anti-i |
|
|
patients with hereditary spherocytosis (HS) have an increased osmotic fragility
|
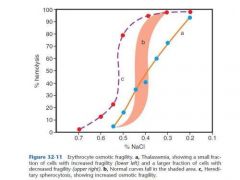
|
|
|
12 yog whose father has HS has an osmotic fragility performed immediately on drawing her blood . the result is within the reference range but at the high (borderline increased fragility) end. a 24 hour incubation test is performed to explore this further. which patient 1 or 2 demonstrates HS?
|

|
|
|
several red cell membrane proteins which defend against the body's own complement system are decreased or absent in PNH. the two key proteins are CD55 and CD59. what are their functions.?
|
CD55: decay accelarating factor, it anatagonizes the convertase complexes of the complement.
CD59: is a membrane inhibitor of reactive lysis (MIRL). it controls and inhibits the membrane complex (C5B-C9) |
|
|
name the three hemoglobins that are normally present in the first trimester of gestation?
|
HbsGower 1, Portland, and Gower 2. these can be roughly thought of as the first attempts at Hbs A,F, ans A2, only using the zeta and epsilon chains which are analogs of the alpha, beta, delta and gamma chains
|
|
|
20 month old has 8% Hb F on electrophoresis. is this diagnostic of a hemoglobinopathy
|
no, about 20% of toddlers 12-24 months have HbF levels 3-10%
|
|
|
after receiving primaquine, a person with which G6PD variant would be likely to have the most severe hemolysis?
variant A variant B Variant Mediterranean |
the mediterannean variant patient may have as little as 1% G6PD activity and is at risk for greater hemolysis than the black person with variant A, who has 5-15% activity. Variant B is the most common of the 300-plus G6PD variants and is normal
|
|
|
heinz bodies may be found in the red cells in the most common red cell enzyme deficiency involving the glycolytic pathway?
|
no. False. Heinz bodies are seen in pyruvate kinase deficiency
|
|
|
phenotypes seen in alpha thalessemia with 1,2,3, and 4 gene deletions??
|
one gene deleted- silent carrier
two genes deleted- alpha thalassemia trait three genes deleted- Hb H disease four genes deleted- hydrops fetalis |
|
|
what 2 hemoglobins migrate with the HbS at alkaline electrophoresis?
at acid pH these two migrate with? |
D, G (and Lepore)
A |
|
|
in cooley's anemia what is the predominant Hb?
|
Hb F is the predominant Hb in the homozygotes for beta-thalassemia. in beta 0- thalessemia, Hb F can be as high as 98%. in beta+-thalessemia, a slightly less debelitating form of homozygosity for beta-thalessemia, Hb F is 60-95%
|
|
|
HbA2 quantitation is most often used to identify individuals with what common hemoglobinopathy?
|
beta-thalassemia trait; HbA2 levels may be up to 7%; 3.5% is usually the high end of the reference range
|
|
|
contrast the result of the acid elution slide test for
Hb F in persons with hereditary persistence of fetal hemoglobin (HPFH) and thalassemia |
in HPFH, Hb F is evenly distributed among the red cells. in thalassemia and other hemoglobinopathies where Hb F is increased, the Hb F is unevenly distributed
|
|
|
give the levels of the following hemoglobins in sickle cell disease
Hb SS Hb AS Hb C Hb SC Hb AC |
Hb SS: S >80%, F 1-2%, A2 2-5%
Hb AS: A 50-65%, S 35-45%, F 0 -1%, A2 2-5% Hb C: C >90%, F 2-4%, A2 2% Hb SC: s 50%, C <50%, F <2%, A2 2% Hb AC: A 55%, C 40%, F <7%, A2 2% |
|
|
homozygotes for Hb D, E and G are usually asymptomatic. What laboratory result is abnormal in one of the hemoglobinopathies, giving a clue to its presence?
|
Hb E is abnormal, persons with both Hb E disease and trait have low MCV. this may suggest thalassemia trait until explored more thouroughly
|
|
|
after the neonatal period , Hb Bart's is seen only in what disorder?
|
this is Hb H disease in which three of the four alpha-globin genes are deleted. these patients have 5-30% Hb H and just a trace of Hb Bart's as adults
|
|
|
are reticulocytes preferentially destroyed in glucose-6 -phosphate dehydrogenase (G6PD) deficiency?
|
no, it is the senescent red cells, with less G6PD activity to begin with, that are the greatest risk of hemolysis
|
|
|
TTP?
|
vWF-cleaving protease present in normal individuals cleaves the ultra-large vWF multimers which are secreted by the endothelial cells. deficiency in this protease results in failure to cleave the multimers, which are highly adhesive and induce platelet clumping throughout the circulation. the gene for coding this enzyme is ADAMTS13
|
|
|
warm autoimmune hemolytic anemia is usually due to an IgG antibody. the most common subclass of IgG is ??, and ?? and ??
|
IgG1, IgG2 and IgG4
|
|
|
physical description of a person with Gaisbock's syndrome?
|
obese male smoker, also hypertensive
|
|
|
disorders of the kidney associated with inappropriate erythropoietin production
|
renal tumors, especilly renal cell carcinoma. renal cysts. hydronephrosis
|
|
|
explain the disparity between the percentage of patients taking methyldopa who have a positive DAT and percentage with hemolytic anemia
|
in addition to causing a positive DAT in up to 1/3 of the patients taking the drug methyldopa also decreases mononuclear phagocytic activity, and this may explain the lower than expected persons with hemolysis
|
|
|
double heterozygotes for the sickle cell mutation and beta-thalessemia have more Hb S than Hb A. this is useful in the distinction of Hb S/beta+- thalassemia from Hb AS. How is the distinction between Hb S disease and Hb S/beta0-thalessemia made?
|
Hb S/beta0-thalessemia double heterozygotes have a lower MCV and MCH and an increased HbA2 due to their thalessemia compared to sickle cell disease
|
|
|
a patient with chronic paroxysmal cold hemoglobin (PCH)...what class of immunoglobin is involved in PCH?
|
biphasic igG (Donath-Landsteiner Ab)
chronic PCH is associated with congenital syphyllis |