![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
49 Cards in this Set
- Front
- Back
|
Anemias |
Conditions in which there are too few erythrocytes or an insufficient volume of erythrocytes in the blood. Anemia is a reduction in the total number of sites in the circulating blood or a decrease in the quality or quantity. |
|
|
Polycythemia |
Conditions in which erythrocyte numbers or volume is excessive |
|
|
Common causes of anemia |
1. Impaired erythrocyte production
2.blood loss, acute or chronic
3. Increased erythrocyte destruction
4. Combination of these three factors |
|
|
Macrocytic-Normochromic anemias |
1. Pernicious anemia/Vitamin B12 deficiency
2. Folate deficiency anemia |
|
|
Macrocytic-Normochromic anemias |
1. Pernicious anemia/Vitamin B12 deficiency
2. Folate deficiency anemia |
|
|
Microcytic-hypochromic anemias |
1. Iron deficiency anemia
2. Sideroblastic anemia
3. Thalassemia |
|
|
Normocytic-Normochromic anemias |
1. Aplastic anemia
2. Post hemorrhagic anemia
3. Hemolytic anemia
4. Sickle cell anemia
5. Anemia of chronic disease |
|
|
Compensation for reduced blood volume in anemia |
Interstitial fluid moves into the intravascular space expanding plasma volume. This maintain adequate volume but the blood viscosity decreases. Blood flows faster and more turbulently causing a hyperdynamic circulatory state. Dilation of arterioles, capillaries, and venules decreases vascular resistance and increases flow. The heart rate and stroke volume increase, in an effort to meet normal oxygen demand. These compensatory mechanisms may lead to heart failure if the anemia is not corrected. |
|
|
Symptoms of anemia |
Increased rate and depth of breathing, increased BPG to increase release of oxygen from hemoglobin, skin mucous membranes lips nailbed and conjunctiva pale, if caused by hemolysis skin may be yellow, skin elasticity can decrease early greying of the hair, nervous system manifestations in B12, abdominal pain/n/v/anorexia, low-grade fever of less than 38.5 Celsius from leukocyte pyrogens release from ischemic tissues. In acute or severe, RAAS |
|
|
Eryptosis (premature death of damaged erythrocytes) |
A common mechanism for cellular loss in individuals with anemias secondary to deficiencies of iron, infections, chronic diseases, genetic diseases, and myelodysplastic syndrome. The process is similar to removal of old or senescent erythrocytes. The erythrocytes lifespan may be decreased by as much as 50%. |
|
|
Macrocytic (megaloblastic) anemias |
Characterized by unusually large stem cells (megaloblasts) in the marrow that mature into erythrocytes that are unusually large in size and thickness and volume. These anemias are the result of defective erythrocyte DNA synthesis, commonly caused by B 12 and folate deficiency. Folate and B 12 are coenzymes that are required for nuclear maturation and DNA synthesis. These defective erythrocytes die prematurely, which decreases their numbers in the circulation, causing anemia. There is asynchronous development which leads to an overproduction of hemoglobin, creating a larger than normal erythrocyte.
Additionally there is an increase in the amount of lactic dehydrogenase, which reflects cellular destruction, and an increase in indirect bilirubin, from the breakdown of heme. These labs are biochemical evidence of ineffective erythropoiesis |
|
|
Pernicious anemia (B12 deficiency anemia) |
Pernicious anemia is caused by vitamin B 12 deficiency which is often associated with the end stage of type A chronic atrophic gastritis, congenital or auto immune. The principal disorder in pernicious anemia is an absence of intrinsic factor, a transporter required for absorption of dietary vitamin B 12, which is essential for nuclear maturation and DNA synthesis in erythrocytes.
Pernicious anemia is also frequently a component of autoimmune conditions. Autoimmune gastritis, type a chronic gastritis, causes gastric atrophy which results from destruction of parietal and zymogenic cells. The parietal and zymogenic cells are destroyed and replaced by mucus containing cells, intestinal metaplasia. Gastric mucosal atrophy in which gastric parietal cells are destroyed, results in a deficiency of all secretions of the stomach: HCl, pepsin, and intrinsic factor. Additionally auto antibodies against intrinsic factor prevent the formation of the B 12-intrinsic factor complex. |
|
|
Clinical manifestations of pernicious anemia/vitamin B 12 deficiency anemia |
Pernicious anemia develops slowly possibly over 20 to 30 years; 60 years of age is the median age at diagnosis. Usually severe by the time treatment is sought. When hemoglobin levels are lowered significantly, 7 to 8 g/dL, classic symptoms of anemia arise. Additionally sore tongue that is smooth and beefy red, paresthesia of feet and fingers, difficulty walking. Hepato- megaly can be present due to right-sided heart failure. Neurological manifestations result from nerve demyelination that may produce neuronal death. And increased prevalence of serum vitamin B 12 deficiency has been reported among individuals with all Alzheimer's disease. |
|
|
DDx for Pernicious anemia |
Cestode infection (tapeworm) Neurological disorders Senility Achlorhydria (absence of hydrochloric acid) Alcoholic fatty liver Alcoholic hepatitis Anemia Atrophic gastritis Bone marrow failure Celiac disease/sprue Cirrhosis Folic acid deficiency Gastric cancer Hemolytic anemia Hyperthyroidism Hypothyroidism Immune thrombocytopenic Purpera (ITP) Iron deficiency anemia Macrocytosis Malabsorption Mega blastic anemia Myeloproliferative disease Tropical sprue Unconjugated hyperbilirubinemia Zollinger-Ellison syndrome |
|
|
Labs for pernicious anemia |
Peripheral blood smear: usually shows a macrocytic anemia with a mild leukopenia and thrombocytopenia.
Indirect bilirubin and lactate dehydrogenase assays: indirect bilirubin level maybe elevated because pernicious anemia is a hemolytic disorder associated with increased turnover of bilirubin. The serum lactate dehydrogenase concentration usually is markedly increased (LD usually an indicator of tissue or cellular damage)
Evaluation of gastric secretions: total gastric secretions are decreased to about 10% of the reference range.
Serum cobalamin: Low in patients with pernicious anemia. However it may be within the reference range in certain patients with other forms of cobalamin deficiency. Can be falsely low in patients who are pregnant, taking oral contraceptives, have multiple myeloma, have transcobalamin I deficiency (TCI), have severe folic acid deficiency, or have taken large doses of ascorbic acid. Of note serum cobalamin levels can be in the low reference range in patients with clinical vitamin B 12 deficiency. In these cases, elevated levels of methylmalonic acid and total homocysteine can confirm the diagnosis of pernicious anemia.
Folic acid: serum folic acid can be helpful for ruling out folic acid deficiency
Methylmalonic acid and Homocystine assays: important confirmatory tests, but are not first-line tests. Elevated serum methylmalonic acid and homocysteine levels are found in patients with pernicious anemia. They are probably the most reliable tests for cobalamin deficiency in patients who do not have a congenital metabolism disorder.
Intrinsic factor antibody assay: intrinsic factor antibodies, type 1 and type 2, occur in 50% of patients with pernicious anemia and are specific for this disorder. They can be used to confirm the diagnosis. Parietal cell antibodies occur in 90% of patients with pernicious anemia. However these antibodies are not specific for pernicious anemia.
Shillings test-no longer available in most medical centers: A radioactive cyanocobalami specimen is drunk and urine radioactivity is monitored
A clinical trial of vitamin B 12: intramuscular administration of 1000 µg of vitamin B 12 can be used as a clinical trial for suspected cobalamin deficiency. Subjectively patients who are cobalamin deficient usually begin to experience a marked sense of well-being within 24 hours after administration. Administration of cobalamin produces a marked reticulocytosis, which reaches its maximum level 5 to 7 days after injection; correction of the anemia occurs in approximately three weeks.
Bone marrow aspiration and biopsy. |
|
|
Treatment of pernicious anemia/B12 deficiency anemia |
1 mg (1000 µg) of B12 (cyanocobalamin or hydroxocobalamin) intramuscularly daily X7 days, then weekly for 4-8 weeks then monthly for life
If the oral route is necessary, patients can take 250 to 1000 µg of vitamin B 12 daily. Even with a total absence of intrinsic factor, about 1% of an oral dose is absorbed, the daily requirement for vitamin B 12 is 1µg per day. |
|
|
Folate (folic acid) deficiency anemia |
Folate is in essential vitamin for RNA and DNA synthesis within the maturing erythrocyte. Folates are coenzymes required for the synthesis of Thymine and purines (adenine and guanine) and the conversion of homocysteine to methionine. Deficient production of thymine, in particular, effects cells undergoing rapid division such as bone marrow cells undergoing erythropoiesis.
Humans are totally dependent on dietary intake to meet the daily requirement of 50 to 200 µg per day of folate. Increased amounts are required for pregnant and lactating females.
Alcohol interferes with folate metabolism in the liver, causing a profound depletion of folate stores. |
|
|
Pathophysiology of folate deficiency anemia |
Impaired DNA synthesis secondary to folate deficiency results in megaloblastic cells with clumped nuclear chromatin. Anemia may result from apoptosis of erythroblasts in the late stages of erythropoiesis.
Folate deficiency is associated with neural tube defect's of the fetus, also associated with coronary artery disease. |
|
|
Clinical manifestations of folate deficiency anemia |
Severe cheilosis (scales and fishers of the lips and corners of the mouth), stomatitis (inflammation of the mouth), and painful ulcerations of the buccal mucosa and tongue, characteristic of burning mouth syndrome.
Can be undiagnosed inflammatory bowel disease, folate deficiency may suppress proliferation of the intestinal mucosal, leading to exacerbation of G.I. damage. |
|
|
Evaluation of folate deficiency anemia |
Decreased folate
Decreased red blood cell folate
Increased homocysteine, but normal methylmalonic acid (unlike B12 deficiency, which has increased in both) |
|
|
Evaluation of folate deficiency anemia |
Decreased folate
Decreased red blood cell folate
Increased homocysteine, but normal methylmalonic acid (unlike B12 deficiency, which has increased in both) |
|
|
Treatment of folic acid deficiency anemia |
Folate 1-5 mg PO daily for 1 to 4 months, or until complete hematologic recovery
It is critical to rule out B 12 deficiency first |
|
|
Three specific disorders of microcytic-hypochromic anemia |
Iron deficiency anemia
Sideroblastic anemia
Thalassemia |
|
|
Three specific disorders of microcytic-hypochromic anemia |
Iron deficiency anemia
Sideroblastic anemia
Thalassemia |
|
|
Iron deficiency anemia, background and information |
Iron deficiency anemia is the most common type of anemia worldwide
At risk individuals include those living in poverty, women of childbearing age, and children
Iron deficiency in children is associated with adverse health related manifestations especially cognitive impairment which maybe irreversible.
Children in developing countries can be affected by chronic parasite infestation's that result in intestinal blood and iron loss that outpace dietary intake. Treatment of Helmont infection results in improvement.
Iron deficiency also occurs in individuals with lead poisoning. Treatment of the iron deficiency is associated with a decrease in lead levels. |
|
|
Iron deficiency anemia: pathophysiology |
Iron deficiency anemia can arise from in adequate dietary intake or excessive blood loss, or a combination of the two.
A second category is a metabolic or functional iron deficiency in which various metabolic disorders lead to either insufficient iron delivery to bone marrow or impaired iron use within the marrow.
The most common cause of iron deficiency anemiain developed countries is pregnancy and chronic blood loss. Other causes include menorrhagia, use of meds that cause G.I. bleeding, surgical procedures that decrease stomach acidity, intestinal transit time, and absorption; and insufficient dietary intake of iron and eating disorders such as pica.
Iron in the form of hemoglobin is in constant demand by the body. Iron is recyclable, therefore, the body maintains a balance between iron that is contained in hemoglobin and iron that is in storage and available for future human hemoglobin synthesis. Blood loss disrupt this balance. |
|
|
Three stages of iron deficiency |
Stage one: the body'a iron stores are depleted. Erythropoiesis proceeds normally with the hemoglobin content of erythrocytes remaining normal.
Stage two: iron transportation to bone marrow is diminished resulting in iron deficient erythropoiesis
Stage three: begins when the small hemoglobin-deficient cells enter the circulation to replace the normal aged erythrocytes that have been removed from the circulation. The manifestations of iron deficiency anemia appear in stage III when there is depletion of iron stores and diminished hemoglobin production. |
|
|
Clinical manifestations of iron deficiency anemia |
Early symptoms nonspecific: fatigue, weakness, shortness of breath, pale earlobes palms and conjunctiva.
The fingernails become brittle, thin, coarsely ridged, and spoon shaped or concave (koilonychia) as a result of impaired capillary circulation, unexplained burning mouth syndrome glossitis, angular stomatitis: dryness and soreness in epithelium at the corners of the mouth, esophageal webbing, hyposalivation |
|
|
Labs for iron deficiency anemia |
Complete blood count: The CBC documents the severity of the anemia. Microcytic and hypochromic erythropoiesis--both the mean corpuscular volume (MCV) and the mean corpuscular hemoglobin concentration (MCHC) have values below the normal range. Often the platelet count is elevated but this elevation normalizes after iron therapy.
Peripheral smear: Direct examination of erythrocytes shows microcytic and hypochromic red blood cells unlike in thalassemia, target cells are not present, and anisocytosis (RBCs of unequal size) and poikilocytosis (RBCs abnormally shaped) are not marked.
Serum iron: low
Serum Ferritin level: low
Total iron binding capacity: elevated
**Low serum iron and ferritin levels with an elevated TIBC are diagnostic of iron deficiency** |
|
|
Treatment of iron deficiency anemia |
1. Identify and eliminate sources of blood loss
2. Oral Ferrous iron: Ferrous sulfate 325 mg orally TID, (lower doses 15 to 20 mg of elemental iron daily maybe as effective and cause fewer side effects). Patients should avoid tea and coffee and may take vitamin see 500 units with the iron pill once daily
3. Parenteral Iron: parental iron is reserved for patients who are unable to absorb oral iron or who have increasing anemia despite adequate PO doses of iron. It has a greater morbidity than oral preparations. I VE options include Ferrous sucrose, Ferrous gluconate, and Ferrous dextran.
|
|
|
Sideroblastic anemia: information and background |
Sideroblastic anemia characterized by anemia verifying in severity caused by a defect in mitochondrial heme synthesis. Sideroblastic anemia is characterized by the presence of ring Sideroblasts within the bone marrow. Ring sideroblasts are erythroblasts that contain iron-laden mitochondria arranged in a perinuclear collar around one third or more of the nucleus. Patients with sideroblastic anemia also have increased levels of iron in their tissues. The blood contains hypochromic erythrocytes, either microcytic or macrocytic depending on the form of the disease. |
|
|
Sideroblastic anemia: Pathophysiology |
Sideroblastic anemias have multiple etiologies but all share the commonality of altered heme synthesis in the erythroid cells in bone marrow. Mitochondrial pathways that have Ferrous iron inserted by the enzyme ferrochelatase are disrupted, and lead to the accumulation of iron in the mitochondria and the characteristic sideroblasts.
Sideroblastic anemia's are either acquired or hereditary.
Acquired sideroblastic anemia, which is the most common, occurs as a primary disorder with no known cause, idiopathic, or is associated with other myeloproliferative or myeloplastic disorders. Another form is described as reversible sideroblastic anemia, which can be caused by alcoholism, drug reactions, copper deficiency, and hypothermia.
Hereditary sideroblastic anemia: rare occurs almost exclusively in males, suggested X linked transmission.
**Differentiation of sideroblastic anemia from idiopathic hemochromatosis needs to be confirmed because both are characterized by tissue iron deposition** |
|
|
Leading known cause of primary acquired sideroblastic anemia |
Myelodysplastic syndrome: a group of disorders of hematopoietic stem cells, with all three stem cell lines demonstrating dysplastic characteristics. Two subsets of Myelodysplastic ringed sideroblasts were identified
Pure Sideroblastic anemia: dysplastic features limited to the erythroid line
Second subset: abnormalities of multiple cell lines including major alterations of neutrophils and platelets |
|
|
Sideroblastic anemia: Clinical manifestations |
The anemia can be moderate to severe, with hemoglobin levels from 4 to 10 g/dL, signs of iron overload (hemochromatosis), mild to moderate enlargement of the spleen and liver, occasionally abnormal skin pigmentation bronze-tinted,
hemosiderosis of cardiac tissue:major life-threatening complication. |
|
|
Where is heme synthesized? |
Mitochondria |
|
|
Where is heme synthesized? |
Mitochondria |
|
|
Where is globin synthesized? |
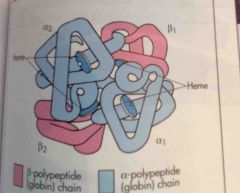
The cytoplasm.
Heme and globin chains (alpha and beta) are manufactured in separate cell compartments, mitochondria and cytoplasm respectively, and then combined in the cytoplasm. |
|
|
Sideroblastic anemia: labs |
CBC: The CBC will show anemia mostly moderate can be severe with a low MCV. Can have dimorphic (normocytic plus microcytic) smears. Siderocytes with Pappenheimer bodies (hypochromic erythrocytes with basophilic iron deposits) are sideroblasts that have matured enough to make it to peripheral blood.
Bone marrow examination: bone marrow is packed with erythrocyte stem cells, and mononuclear phagocytes in the marrow are loaded with iron in the form of Homosiderin. The presence of sideroblasts confirms the diagnosis.
Serum Iron increased with normal TIBC, increased ferritin |
|
|
Sideroblastic anemia: treatment |
Removal of toxic/causative agent's
Pyridoxinee, vitamin B6, therapy: deserves a trial in all cases of sideroblastic anemia. Dosage of B6 that will maintain the hemoglobin level yet prevent toxicity (peripheral neuropathy); typically 50 to 200 mg/dL of vitamin B6. For those who respond treatment is life long.
Supportive blood transfusions for those with sideroblastic anemia that does not respond to pyridoxine (B6) therapy. Must monitor for iron overload which can be fatal.
If iron overload occurs deferoxamine (desferrioxamine; Desferal), an iron chelating agent, can be used (given subQ via pump several hours per day). Deferasirox (Exjade) is an oral iron chleating agent that can be taken PO daily. |
|
|
Normocytic-Normochromic anemias: 5 distinct groups |
1. Aplastic: damage to bone marrow erythropoiesis 2. Post-hemorrhagic: acute blood loss 3. Acquired hemolytic: immune destruction of erythrocytes 4. Hereditary hemolytic: such as sickle cell, destruction by eryptosis 5. Anemia of chronic inflammation |
|
|
Aplastic anemia (normocytic-normochromic anemia): information and background |
Aplastic anemia is a critical condition characterized by pancytopenia, a reduction or absence of all three blood cell types, resulting from failure or suppression of bone marrow to produce adequate amounts of blood cells. Aplastic anemia is relatively rare. The most common type is idiopathic aplastic anemia (primary acquired) which is an autoimmune. Secondary aplastic anemia is caused by a variety of known chemical agents and ionizing radiation. Liver disease (seronegative hepatitis) is also recognized as a cause of aplastic anemia.
Other causes of aplastic anemia: total body irradiation, infection particularly with viruses (HIV, Ebstein-bar, hepatitis, persistent parvovirus B19).
Pure red cell aplasia: another condition associated with aplastic anemia, in which only the erythrocytes are affected. This is a rare disorder and associated with autoimmune, viral, neoplastic (leukemia), Infiltrative disorders of the bone marrow (myelofibrosis), renal failure, hepatitis, mononucleosis, systemic lupus erythematosus. |
|
|
Aplastic anemia: pathophysiology |
The characteristic lesion of aplastic anemia is a hypo-cellular bone marrow that has been replaced with fat. Most cases of idiopathic aplastic anemia result from an autoimmune disease directed against hematopoietic stem cells. |
|
|
Aplastic anemia: clinical manifestations |
The onset of symptoms is insidious and related to the rapidity with which the bone marrow is destroyed and replaced
Approximately 50% of aplastic anemia cases progress rapidly, with a high degree of death from overwhelming infection or bleeding
Initial symptoms depend on which cell line is affected.
Rapidly progressing disease: hypoxemia, pallor, weakness, fever and dyspnea, rapidly developing signs of hemorrhage if platelets are affected.
In both rapid and slow onset aplastic anemia, diminished leukocyte production may result in progressive frequency and prolongation of infection.
Late manifestations: ulcerations of the mouth and pharynx, a low-grade cellulitis in the neck.
Hypoplastic anemia: the rate of decline is slow and the individual may adapt progressively to a new level of hematologic function. This is referred to as hypoplastic anemia rather than aplastic anemia. |
|
|
Aplastic anemia: evaluation/labs |
Blood tests: circulating erythrocytes, leukocytes, and platelets are diminished.
The diagnosis of aplastic anemia is confirmed by a bone marrow biopsy. The bone marrow usually has reduced cellularitu and contains yellowish white material consisting mainly of fat, fibrous tissue, and lymphocytes. Pancytopenia is usually characterized by decreased stem cell and progenitor cell populations to approximately 1% or less of normal |
|
|
Aplastic anemia: treatment |
Bone marrow transplant, immunosuppression, identification of high-risk individuals.
Bone marrow and peripheral blood stem cell transplant from histocompatible siblings: often cures the underlying bone marrow failure
Immunosuppression: remains the treatment of choice for those unable to obtain bone marrow transplant. Antithymocyte globulin (ATG): specifically suppresses lymphocytes, including those auto reactive lymphocytes destroying the bone marrow cells. Cyclosporine: often used in combination with ATG, broadly suppresses the activity of immune cells. Cyclosporine plus ATG has increased the response and survival rates to as much as 70 to 80%. Tacrolimus: another immuno suppressant. Prograf. Corticosteroids: often used concurrently with ATG and cyclosporine
Recombinant hematopoietic growth factors: granulocyte-macrophage colony stimulating factor (GM-CSF), IL-6, epoetin have been shown to be helpful in both children and adults |
|
|
Post hemorrhagic, acute blood loss anemia (normocytic normochromic anemia) |
Caused by acute blood loss. A normal healthy young adult can tolerate a blood loss of 500 to 1000 mL (10 to 20% volume), without experiencing any symptoms. When blood loss exceeds 1500 mL the symptoms are apparent even in recumbent position. After acute blood loss, lost plasma is replaced by mobilizing water and electrolytes from tissues and interstitial spaces to the vascular system. The hemodilution that results lowers the hematocrit value; concurrently there is often a rapid elevation in circulating neutrophils and platelets. Reduced tissue oxygenation stimulates production of erythropoietin and increasing production of erythrocytes (reticulocytes) in the bone marrow.
Initial treatment: restoration of blood volume by IV saline, dextran, albumin, or plasma. Large volume loss may require blood transfusion.
Successful therapy is first indicated by a return of erythrocytes to their normal size and shape. As the bone marrow begins to produce more erythrocytes, an increase in the number of reticulocytes 10% to 15% after seven days is seen. A normal erythrocyte count is usually noted in 4 to 6 weeks, but hemoglobin restoration may take 6 to 8 weeks. |
|
|
Hemolytic anemia (normocytic normal chromic anemia):background and information |
The predominant event in hemolytic anemia's is premature accelerated distraction of erythrocytes, either episodically or continuously. The consequences of the anemia are elevated levels of erythropoietin to induce accelerated production of erythrocytes and an increase in the products of hemoglobin catabolism. Hemolytic anemia's maybe either congenital or acquired. |
|
|
Haptoglobin |
Haptolobin is an acute-phase reactant whose principal clinical utility is in defining conditions of hemolysis. Levels can also become elevated in infection and inflammation. The reference range of haptoglobin in adults is 30 to 200 mg/dL.
Haptoglobin is a colorless protein that transports "freed" hemoglobin released from destroyed red blood cells. It is produced by the liver and collects the hemoglobin from destroyed red blood cells, then transports it back to the liver, where heme is converted to bilirubin. In the setting of increased red blood cell destruction, haptoglobin becomes depleted, and the free hemoglobin dimmers are filtered by the kidney, ultimately producing hemosiderin. Macrophages destroy the hemoglobin-haptoglobin complexes. The hemoglobin-haptoglobin complex is removed from the bloodstream within minutes.
Is used as an acute-phase marker of red blood cell distruction. It's value decreases and may even be absent when RBCs are destroyed at twice the normal rate. Decreased levels of haptoglobin have 83% sensitivity and 96% specificity for hemolytic anemia. Overall a serum haptoglobin level below 25 mg/dL equates to an 87% probability of predicting hemolytic disease.
Conditions with increased haptoglobin: elevated ESR diseases, obstructive or biliary diseases, steroid use, aplastic anemia, diabetes, smoking, nephrotic syndrome, increased estrogen level
Decreased or absent haptoglobin levels: intravascular hemolysis, extravascular hemolysis, intramedullary hemolysis, genetic, haptoglobin absent in 1% of whites and 4 to 10% of blacks, cirrhosis, infancy, pregnancy, burns.
|
|
|
Names of increased cell lines (myeloproliferative) |
Polycythemia: Excessive red blood cell production
Erythrocytosis: increased erythrocytes
Leukocytosis: increased levels of white blood cells
Thrombocytosis: increased levels of platelets |