Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
421 Cards in this Set
- Front
- Back
|
Aspartate
|
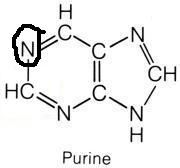
What molecule contributed this atom?
|
|
|
Carbon Dioxide
|
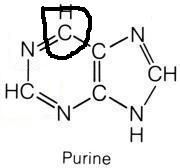
What molecule contributed this atom?
|
|
|
Glutamine
|
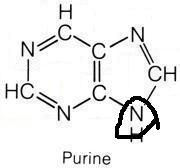
What molecule contributed this atom?
|
|
|
Glutamine
|
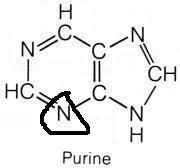
What molecule contributed this atom?
|
|
|
Glycine
|
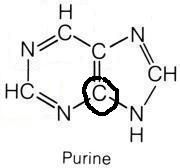
What molecule contributed this atom?
|
|
|
Glycine
|
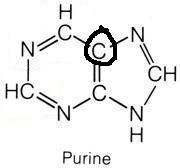
What molecule contributed this atom?
|
|
|
Glycine
|
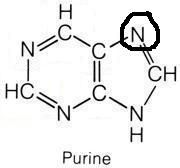
What molecule contributed this atom?
|
|
|
N10-formyl-tetrahydrofolate
|
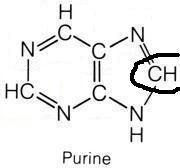
What molecule contributed this atom?
|
|
|
N10-formyl-tetrahydrofolate
|
What molecule contributed this atom?
|
|
|
Aspartate
|
What molecule contributed this atom?
|
|
|
Aspartate
|
What molecule contributed this atom?
|
|
|
Aspartate
|
What molecule contributed this atom?
|
|
|
Aspartate
|
What molecule contributed this atom?
|
|
|
Carbamoyl Phosphate
|
What molecule contributed this atom?
|
|
|
Carbamoyl phosphate
|
What molecule contributed this atom?
|
|
|
What does it mean for genetic code to be commaless?
|
Read from a fixed starting point as a continuous sequence of bases
|
|
|
What does it mean for genetic code to be non-overlapping?
|
Read from a fixed starting point
|
|
|
What does it mean for genetic code to be universal?
|
Genetic code is conserved throughout evolution
|
|
|
What are the properties of the genetic code?
|
1. Unambiguous.
2. Degenerate/redundant. 3. Commaless/nonoverlapping 4. Universal |
|
|
When is genetic code not commaless/nonoverlapping?
|
In some viruses
|
|
|
What are exceptions to universality of genetic code?
|
1. Mitochondria
2. Archaebacteria 3. Mycoplasma 4. Some yeasts |
|
|
Name that mutation: Same amino acid, often with a base change in 3rd position of codon
|
Silent mutation
|
|
|
What kind of mutation is called: silent
|
Same amino acid, often with a base change in 3rd position of codon
|
|
|
What mutation is masked by tRNA wobble?
|
Silent mutations
|
|
|
Name that mutation: Changed amino acid whose structure is dissimilar to proper amino acid
|
Missense mutation (not conservative)
|
|
|
Name that mutation: Changed amino acid whose structure is similar to proper amino acid
|
Conservative missense mutation
|
|
|
What kind of mutation is called: missense
|
Amino acid is changed. If the structure of the new amino acid is similar to the original, it is called conservative.
|
|
|
Name that mutation: Change resulting in early stop codon
|
Nonsense mutation
(Mnemonic: Stop the nonsense!) |
|
|
What kind of mutation is called: nonsense
|
Change resulting in early stop codon
(Mnemonic: Stop the nonsense!) |
|
|
Name that mutation: change resulting in misreading of all nucleotides downstream, usually resulting in a truncated protein
|
Frame shift mutation
|
|
|
What kind of mutation is called: frameshift
|
change resulting in misreading of all nucleotides downstream, usually resulting in a truncated protein
|
|
|
Mutations ordered by decreasing severity of damage
|
1. Nonsense
2. Missense 3. Silent |
|
|
Eukaryotic genome: single/multiple origins of replication
|
multiple
|
|
|
Prokaryotic genome: single/multiple origins of replication
|
single
|
|
|
Eukaryotic genome: Trigger for replication
|
Consensus sequence of AT-rich base pairs
|
|
|
Prokaryotic genome: Describe DNA replication
|
Continuous bidirectional DNA synthesis on leading strand and discontinuous (Okazaki fragments) on lagging strand
|
|
|
Enzyme function: DNA topoisomerases
|
Create a nick in the helix to relieve supercoils
|
|
|
DNA Topoisomerase I: Mechanism
|
cuts one strand, passes the other through it then reanneals the cut strand
|
|
|
DNA Topoisomerase II: Mechanism
|
cuts both strands, and passes an unbroken double strand through it then reanneals the cut strand
|
|
|
Enzyme function: Primase
|
Makes an RNA primer on which DNA polymerase III can initiate replication
|
|
|
DNA polymerase III: Mechanism
|
1. Adds deoxynucleotides to the 3' end until it reaches primer of preceding fragment
2. 3' to 5' exonuclease activity "proofreads" each added nucleotide |
|
|
DNA polymerase III: Which direction does it read?
|
3' to 5'
|
|
|
DNA polymerase III: Which direction does it write?
|
5' to 3'
|
|
|
DNA polymerase III: Which direction does it proofread?
|
3' to 5'
|
|
|
Enzyme function: DNA polymerase III
|
Elongates the chain
|
|
|
Enzyme function: DNA polymerase I
|
Degrades RNA primer and fills in the gap with DNA
|
|
|
DNA polymerase I: Which direction does it read?
|
3' to 5'
|
|
|
DNA polymerase I: Which direction does it write?
|
5' to 3'
|
|
|
DNA polymerase I: Which direction does it proofread?
|
5' to 3'
|
|
|
Enzyme function: DNA helicase
|
Separates the two strands of DNA into single strands allowing for replication to occur. The position of these separated strands is called the replication fork.
|
|
|
Types of DNA repair
|
Single stranded:
1. Nucleotide excision repair 2. Base excision repair 3. Mismatch repair Double stranded: 1. Nonhomologous end joining |
|
|
Nucleotide excision repair: Mechanism
|
1. Specific endonucleases release the oligonucleotide containing damaged bases
2. DNA polymerase and ligase fill and reseal the gap, respectively |
|
|
In what condition is nucleotide excision repair mutated?
|
Xeroderma pigmentosa (dry skin with melanoma and other cancers)
|
|
|
Base excision repair: Mechanism
|
1. Specific glycosylases recognize and remove damaged bases
2. AP endonuclease cuts DNA at apyrimidinic site 3. Empty sugar is removed 4. Gap is refilled and resealed |
|
|
Mismatch repair: Mechanism
|
1. Unmethylated, newly synthesized string is recognized
2. Mismatched nucleotides are removed 3. Gap is refilled and resealed |
|
|
In what condition is mismatch excision repair mutated?
|
Hereditary Nonpolyposis Colon Cancer
|
|
|
Nonhomologous end joining: Mechanism
|
Brings together two ends of DNA fragments (no requirement for homology)
|
|
|
What is on the 5' end of a nucleotide
|
Triphosphate
|
|
|
What is on the 3' end of a nucleotide
|
Hydroxyl group
|
|
|
True/False: DNA is synthesized 5' to 3'
|
True
|
|
|
True/False: DNA is synthesized 3' to 5'
|
False
|
|
|
True/False: RNA is synthesized 5' to 3'
|
True
|
|
|
True/False: RNA is synthesized 3' to 5'
|
False
|
|
|
True/False: Protein synthesis proceeds 5' to 3'
|
True
|
|
|
True/False: Protein synthesis proceeds 3' to 5'
|
False
|
|
|
Types of RNA and their important qualities
|
Massive, Rampant, Tiny
mRNA is the largest type rRNA is the most abundant type tRNA is the smallest type |
|
|
What does eukaryotic RNA polymerase I make?
|
rRNA
|
|
|
What does eukaryotic RNA polymerase II make?
|
mRNA
|
|
|
What does eukaryotic RNA polymerase III make?
|
tRNA
|
|
|
Which RNA polymerase makes rRNA?
|
eukaryotic RNA polymerase I and prokaryotic RNA polymerase
|
|
|
Which RNA polymerase makes mRNA?
|
eukaryotic RNA polymerase II and prokaryotic RNA polymerase
|
|
|
Which RNA polymerase makes tRNA?
|
eukaryotic RNA polymerase III and prokaryotic RNA polymerase
|
|
|
True/False: RNA polymerase proofreads.
|
False
|
|
|
True/False: RNA polymerase does not proofread.
|
True
|
|
|
Special points about RNA polymerase II
|
1. Opens DNA at promoter site
2. Inhibited by alpha-amanitin |
|
|
What does alpha-amanitin do?
|
Inhibits RNA polymerase II leading to hepatic necrosis
|
|
|
mRNA initiation codons
|
1. AUG (inAUGurates protein synthesis)
2. GUG (rarely) |
|
|
What does the mRNA initiation codon code for?
|
Methionine in eukaryotes. formyl-methionine in prokaryotes.
|
|
|
mRNA stop codons
|
1. UGA (U Go Away)
2. UAA (U Are Away) 3. UAG (U Are Gone) |
|
|
Define promoter of gene expression.
|
Site where RNA polymerase and multiple other transcription factors bind to DNA upstream from gene locus
|
|
|
What characterizes a promoter of gene expression?
|
AT-rich upstream sequence with TATA and CAAT boxes
|
|
|
What is the result of promoter mutation?
|
Dramatic decrease in amount of gene transcribed
|
|
|
Define enhancer of gene expression.
|
Stretch of DNA that alters gene expression by binding transcription factors. May be located close to, far from, or even within the gene whose expression it regulates.
|
|
|
Define operator of gene expression
|
Site where repressors bind
|
|
|
What is alternative splicing?
|
Rearrangement of exons to make unique proteins
|
|
|
What is the sequence of mRNA splicing?
|
1. Primary transcript combines with snRNP ("snerp") to form spliceosome
2. Lariat-shaped intermediate is generated 3. Lariat is released to remove intron precisely and join two exons |
|
|
Where and when does eukaryotic RNA processing happen?
|
In the nucleus after transcription
|
|
|
What is the initial RNA transcript called?
|
heterogeneous nuclear RNA (hnRNA)
|
|
|
What are the steps in processing hnRNA to make mRNA? (Note: This is more than splicing.)
|
1. Capping on 5' end with 7-methyl-G
2. Polyadenylation on 3' end (approximately 200 As) 3. Splicing out of introns |
|
|
How many nucleotides does tRNA contain?
|
75 to 90 nucleotides
|
|
|
What sequence does every tRNA share at the 3' end?
|
CCA along with a high percentage of chemically modified bases
|
|
|
Amino acid binding to tRNA: Where (on the tRNA) and how?
|
Where: 3' end
How: Covalently |
|
|
What is the enzyme involved in processing tRNA
|
Aminoacyl tRNA synthetase (uses 1 ATP)
|
|
|
Aminoacyl tRNA synthetase: Mechanism
|
1. Scrutinizes amino acid before it binds to tRNA
2. Binds AMP-amino group to 3' end of tRNA 3. Scrutinizes amino acid again. If incorrect, bond is hydrolyzed. |
|
|
What is wrong with a mischarged tRNA
|
Reads the regular bond but inserts wrong amino acid.
|
|
|
Which position on the codon is the wobble position?
|
3rd position
|
|
|
Names of the steps in protein synthesis
|
1. Initiation
2. Elongation 3. Termination |
|
|
Sequence of events in the initiation step of protein synthesis.
|
1. Initiation factors assemble the 40S ribosomal subunit with the initiator tRNA
2. mRNA and (60S?) ribosomal subunit combine with the 40S subunit. 3. Initiation factors are released. |
|
|
Sequence of events in the elongation step of protein synthesis.
|
1. Aminoacyl tRNA binds to the A site
2. Peptidyltransferase catalyzes peptide bond formation. 3. Peptidyltransferase transfers growing polypeptide to amino acid in A site. 4. Ribosome advances three nucleotides toward 3' end of RNA moving peptidyl RNA to P site. |
|
|
Sequence of events in the termination step of protein synthesis.
|
1. Completed protein is released from ribosome.
2. Ribosome dissociates. |
|
|
Role of ATP in protein synthesis
|
ATP does tRNA Activation (charging)
|
|
|
Role of GTP in protein synthesis
|
GTP does tRNA Going places (aka translocation) and Gripping
|
|
|
Role of A site in protein synthesis
|
A site holds incoming Aminoacyl tRNA.
|
|
|
Role of P site in protein synthesis
|
P site accomodates growing Peptide.
|
|
|
Role of E site in protein synthesis
|
E site holds Empty tRNA as it Exits
|
|
|
Which post-translational modification involves removal of N or C terminal pro-peptides from zymogens to generate mature proteins?
|
Trimming
|
|
|
What happens in post-translational trimming?
|
removal of N or C terminal pro-peptides from zymogens to generate mature proteins
|
|
|
Which post-translational modification involves phosphorylation?
|
post-translational covalent alteration
|
|
|
What happens during post-translational covalent alterations?
|
Either:
1. Phosphorylation 2. Glycosylation 3. Hydroxylation |
|
|
Which post-translational modification involves glycosylation?
|
post-translational covalent alteration
|
|
|
Which post-translational modification involves hydroxylation?
|
post-translational covalent alteration
|
|
|
What happens during proteasomal degradation?
|
Attachment of ubiquitin to defective proteins to tag them for breakdown.
|
|
|
Ubiquitin or Ubiquinone: Proteosomal degradation
|
Ubiquitin
|
|
|
Ubiquitin or Ubiquinone: Coenzyme Q in oxidative phosphorylation
|
Ubiquinone
|
|
|
Where in the cell does the following occur: Fatty acid oxidation (beta-oxidation)
|
Mitochondria
|
|
|
Where in the cell does the following occur: acetyl-CoA production
|
Mitochondria
|
|
|
Where in the cell does the following occur: Krebs cycle
|
Mitochondria
|
|
|
Where in the cell does the following occur: Glycolysis
|
Cytoplasm
|
|
|
Where in the cell does the following occur: Fatty acid synthesis
|
Cytoplasm
|
|
|
Where in the cell does the following occur: Hexose Monophosphate Shunt
|
Cytoplasm
|
|
|
Where in the cell does the following occur: Protein Synthesis
|
Rough endoplasmic reticulum in the cytoplasm
|
|
|
Where in the cell does the following occur: Steroid synthesis
|
Smooth endoplasmic reticulum in the cytoplasm
|
|
|
Where in the cell does the following occur: Gluconeogenesis
|
Pathway has steps in the mitochondria and in the cytoplasm
|
|
|
Where in the cell does the following occur: Urea cycle
|
Pathway has steps in the mitochondria and in the cytoplasm
|
|
|
Where in the cell does the following occur: Heme synthesis
|
Pathway has steps in the mitochondria and in the cytoplasm
|
|
|
What type of bonds hold the phosphoryls together in ATP, and how much energy are the bonds worth?
|
Phosphoanhydride bonds are worth 7 kilocalories per mole (but only between the alpha and beta and the beta and the gamma, thus AMP's phosphoryl is not cleaved off for energy)
|
|
|
How many ATP molecules are produced by aerobic metabolism of glucose?
|
38 via the Malate shuttle, and 36 via the G3P shuttle.
|
|
|
In aerobic metabolism of glucose, which pathway produces 38 ATP?
|
Malate shuttle
|
|
|
In aerobic metabolism of glucose, which pathway produces 36 ATP?
|
G3P shuttle
|
|
|
How much ATP is produced by anaerobic glycolysis?
|
2 ATP per glucose
|
|
|
What is this molecule an activated carrier of?: ATP
|
Phosphoryls
|
|
|
What is this molecule an activated carrier of?: NADH
|
Electrons
|
|
|
What is this molecule an activated carrier of?: NADPH
|
Electrons
|
|
|
What is this molecule an activated carrier of?: FADH2
|
Electrons
|
|
|
What is this molecule an activated carrier of?: Coenzyme A
|
Acyl
|
|
|
What is this molecule an activated carrier of?: Lipoamide
|
Acyl
|
|
|
What is this molecule an activated carrier of?: Biotin
|
CO2
|
|
|
What is this molecule an activated carrier of?: Tetrahydrofolate
|
1-carbon units
|
|
|
What is this molecule an activated carrier of?: S-adenosyl-methionine
|
Methyl groups
|
|
|
What is this molecule an activated carrier of?: Thiamine Pyrophosphate
|
Aldehydes
|
|
|
What activated carriers carry: Phosphoryl
|
ATP and GTP
|
|
|
What activated carriers carry: Electrons
|
1. NADH
2. NADPH 3. FADH2 |
|
|
What activated carriers carry: Acyl
|
1. Coenzyme A
2. Lipoamide |
|
|
What activated carriers carry: CO2
|
Biotin
|
|
|
What activated carriers carry: 1-carbon units
|
1. Tetrahydrofolates (originally as formyl then methyl)
2. Biotin (as CO2) 3. S-adenosyl-methionine (as CH3) |
|
|
What activated carriers carry: CH3 groups
|
1. S-adenosyl-methionine
2. N5-methyl-THF |
|
|
What activated carriers carry: Formyl groups
|
N10-formyl-THF
|
|
|
What activated carriers carry: Aldehydes
|
Thiamine Pyrophosphate
|
|
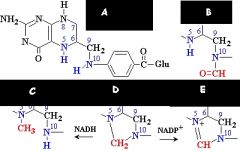
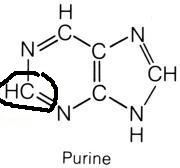
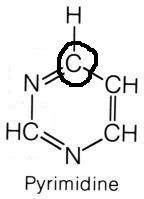
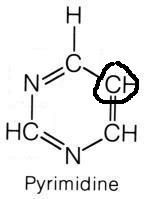
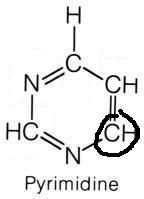
What are the names of these structures?
|
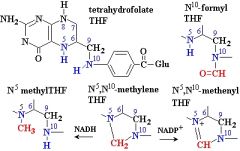
.
|
|
|
ATP and methionine react to form what?
|
S-adenosyl-methionine
|
|
|
What reacts to yield S-adenosyl-methionine?
|
ATP and methionine
|
|
|
What vitamin is necessary for regeneration of S-adenosyl-methionine?
|
Vitamin B12
|
|
|
When is NAD used?
|
Catabolic processes to carry reducing equivalents away as NADH
|
|
|
When is NADPH used?
|
1. Anabolic process (steroid and fatty acid synthesis)
2. Respiratory burst 3. P-450 |
|
|
Where does NADPH come from?
|
HMP shunt
|
|
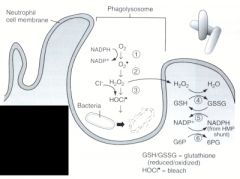
Name the enzymes used in the oxygen-dependent respiratory burst.
|
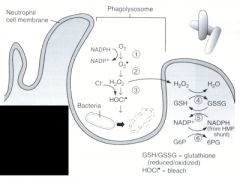
1. NADPH oxidase
2. Superoxide dismutase 3. Myeloperoxidase 4. Catalase/Glutathione peroxidase 5. Glutathione reductase 6. Glucose-6-phosphate dehydrogenase |
|
|
What disease results from NADPH oxidase deficiency?
|
Chronic Granulomatous Disease
|
|
|
This enzyme phosphorylates glucose with high affinity.
|
Hexokinase (as opposed to glucokinase)
|
|
|
This enzyme phosphorylates glucose with low affinity.
|
Glucokinase (as opposed to hexokinase)
|
|
|
This enzyme phosphorylates glucose with a low capacity.
|
Hexokinase (as opposed to glucokinase)
|
|
|
This enzyme phosphorylates glucose and is feedback inhibited by Glucose-6-Phosphate.
|
Hexokinase (as opposed to glucokinase)
|
|
|
This enzyme phosphorylates glucose with a high capacity.
|
Glucokinase (as opposed to hexokinase)
|
|
|
This enzyme phosphorylates glucose and is not feedback inhibited.
|
Glucokinase (as opposed to hexokinase)
|
|
|
Glucokinase: Where is it found and why does it do what it does?
|
Found in the liver and pancreatic beta cells. Phosphorylates glucose to sequester it after a big meal.
|
|
|
Hexokinase: Where is it found and why does it do what it does?
|
Found in every cell's cytoplasm. Phosphorylates glucose to proceed with glycolysis.
|
|
|
What are the net reactants and products in glycolysis.
|
Reactants
1. Glucose 2. 2 Phosphates 3. 2 ADP 4. 2 NAD Products 1. 2 Pyruvate 2. 2 ATP 3. 2 NADH 4. 2 H+ 5. 2 H20 |
|
|
What are the rate limiting steps of glycolysis?
|
1. Hexokinase (Glucose to Glucose-6-P)
2. *Phosphofructokinase-1 (Fructose-6-P to Fructose-1,6-BP) 3. Pyruvate kinase (Phosphoenolpyruvate to Pyruvate) |
|
|
Phosphofructokinase-1: What does it do, and what stimulates and inhibits it?
|
PFK-1 1-phosphorylates fructose-6-phosphate to produce Fructose-1,6-Bisphosphate.
Inhibited by: 1. ATP (don't need more of me) 2. Citrate (my cycle is going well) Stimulated by: 1. AMP (Hey, we need more ATP) 2. Fructose-2,6-BP (The fact that I'm being made means there's tons of glucose.) |
|
|
Pyruvate kinase: What does it do, and what stimulates and inhibits it?
|
Pyruvate kinase converts phosphoenolpyruvate to pyruvate, thereby producing two ATP.
Inhibited by: 1. ATP (don't need more of me) 2. Alanine (I came from pyruvate, so we don't need any more.) Stimulated by: 1. Fructose-1,6-BP (I was told we needed more ATP, so here I am, so you better move the line along.) |
|
|
Pyruvate dehydrogenase: What does it do, and what stimulates and inhibits it?
|
Pyruvate dehydrogenase converts pyruvate to acetyl-coA, and produces NADH and CO2.
Stimulated by: excess pyruvate? Inhibited by: 1. NADH (Listen, seriously, we don't need anymore of me.) 2. NADH (You produce NADH, soon there'll be more of me.) 3. Acetyl-CoA (Enough of me, save your pyrvuate.) |
|
|
Draw a diagram showing the relationship between:
1. glucose 2. fructose-6-P 3. insulin 4. glucagon 5. PFK-1 6. PFK-2 7. F-1,6-BPAse 8. F-2,6-BPAse 9. Fructose-1,6-Bisphosphate 10. Fructose-2,6-Bisphosphate |
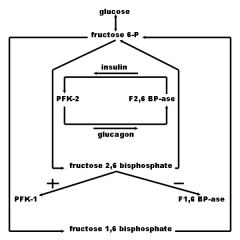
from Rumors Were True blog
|
|
|
What disease state is glycolytic enzyme deficiency generally associated with?
|
Hemolytic anemia
|
|
|
What is the mechanism of hemolytic anemia in someone with glycolytic enzyme deficiency?
|
1. Lack of glycolysis leads to lack of ATP in RBCs
2. Lack of ATP leads to inactivity of Na, K-ATPase pump. 3. Lack of the pump leads to sodium influx. 4. Water follows sodium into the cell. 5. The cell swells and bursts. |
|
|
What are the two most common glycolytic enzyme deficiencies?
|
Pyruvate kinase (95% of cases) followed by glucose phosphate isomerase (4% of cases)
|
|
|
What are the 5 cofactors necessary for pyrvuate dehydrogenase?
|
Lipoic acid plus the first four B vitamins in their active forms:
1. B1: TPP 2. B2: FAD 3. B3: NAD 4. B5: CoA |
|
|
What are the 5 cofactors necessary for alpha-ketoglutarate dehydrogenase?
|
Lipoic acid plus the first four B vitamins in their active forms:
1. B1: TPP 2. B2: FAD 3. B3: NAD 4. B5: CoA |
|
|
What are the net reactants and products in the reaction that Pyruvate Dehydrogenase catalyzes?
|
Reactants:
1. Pyruvate 2. CoA 3. NAD Products 1. Acetyl CoA 2. CO2 3. NADH |
|
|
What activates and what inhibits pyruvate dehydrogenase?
|
Activated by exercise, which stimulates:
1. Increased NAD/NADH ratio (We need more NADH.) 2. Increased ADP (We need more ATP.) 3. Ca2+ (More of me leads muscles to contract, and I'm taken up by mitochondria where I tell PDH that we need more ATP.) Inhibited by: 1. NADH (No more of me please) 2. ATP (likewise) 3. Acetyl CoA (ditto) |
|
|
Lipoamide or lipoate: Which carries aldehydes?
|
Lipoamide
|
|
|
Lipoamide or lipoate: Which is a cofactor for pyruvate dehydrogenase?
|
Lipoate (Lipoic acid)
|
|
|
What toxin inhibits lipoic acid?
|
Arsenic
|
|
|
What is the presentation of arsenic toxicity?
|
1. Vomiting
2. Rice water stools 3. Garlic breath |
|
|
Pyruvate dehydrogenase deficiency: Mechanism
|
Backup of pyruvate and alanine leads to lactic acidosis.
|
|
|
Pyruvate dehydrogenase deficiency: Congenital or Acquired
|
Both. Acquired cases happen in cases of B1 deficiency (such as in alcoholics.)
|
|
|
Pyruvate dehydrogenase deficiency: Presentation
|
Lactic acidosis and neurologic defects
|
|
|
Pyruvate dehydrogenase deficiency: Treatment
|
Increased intake of ketogenic nutrients (such as high fat content or increased lysine and leucine)
|
|
|
What are the miscellaneous fates of pyruvate, and what are the end products used for?
|
1. Alanine: Carries amino groups to the liver from muscle
2. Oxaloacetate: Replenishes TCA cycle or is used gluconeogenesis 3. Acetyl-CoA: Used in TCA cycle 4. Lactate: No good use |
|
|
Which tissues and organs primarily convert pyruvate into lactate?
|
1. RBCs and WBCs
2. Lens and cornea 3. Renal medulla 4. Testes |
|
|
What enzymes and cofactors are used in conversion of pyruvate to alanine?
|
Enzyme: Alanine Transaminase (ALT)
Cofactors: None |
|
|
What enzymes and cofactors are used in conversion of pyruvate to oxaloacetate?
|
Enzyme: Pyruvate Carboxylase (contains biotin and magnesium)
Cofactors: CO2 and ATP |
|
|
What are the reactants and products in the reaction catalyzed by pyruvate carboxylase?
|
Reactant:
Pyruvate (with CO2 and ATP) Product: Oxaloacetate |
|
|
What are the reactants and products in the reaction catalyzed by lactate dehydrogenase?
|
This reaction is reversible, so the products can switch with the reactants.
Reactants: 1. Pyruvate 2. NADH (rehydrogenates in this direction) 3. H+ Products: 1. Lactate 2. NAD |
|
|
Where do the various pyruvate transformation reactions happen?
|
Cytosol:
1. ALT (Alanine to/from pyruvate) 2. LDH (Lactate to/from pyruvate) Mitochondria 1. Pyruvate carboxylase (pyruvate to oxaloacetate) 2. Pyruvate dehydrogenase (pyruvate to acetyl-coa) |
|
|
Where does the Cori Cycle happen?
|
In the liver and muscle/RBCs
Liver: Pyruvate converts to glucose Muscle/RBCs: Glucose converts to Pyruvate |
|
|
What is the purpose of the Cori cycle?
|
Transfers excess reducing equivalents from RBCs and the muscle to liver so they can function anaerobically
|
|
|
What reaction does citrate synthase catalyze?
|
Oxaloacetate and acetyl coA combine to yield citrate.
|
|
|
What is the order of the citric acid cycle beginning at citrate?
|
CAn I Keep Selling Sex For Money, Officer?
1. Citrate 2. cis-Aconitate 3. Isocitrate 4. alpha-Ketoglutarate 5. Succinyl CoA 6. Succinate 7. Fumarate 8. Malate 9. Oxaloacetate |
|
|
What is the order of the citric acid cycle beginning at cis-aconitate?
|
1. cis-Aconitate
2. Isocitrate 3. alpha-ketoglutarate 4. succinyl coA 5. succinate 6. fumarate 7. money 8. oxaloacetate 9. citrate |
|
|
What is the order of the citric acid cycle beginning at isocitrate?
|
1. isocitrate
2. alpha-ketoglutarate 3. succinyl coa 4. succinate 5. fumarate 6. malate 7. oxaloacetate 8. citrate 9. cis-aconitate |
|
|
What is the order of the citric acid cycle beginning at alpha-ketoglutarate?
|
1. alpha-ketoglutarate
2. succinyl coA 3. succinate 4. fumarate 5. malate 6. oxaloacetate 7. citrate 8. cis-aconitate 9. isocitrate |
|
|
What is the order of the citric acid cycle beginning at succinyl coA?
|
1. succinyl coA
2. succinate 3. fumarate 4. malate 5. oxaloacetate 6. citrate 7. cis-aconitate 8. isocitrate 9. alpha-ketoglutarate |
|
|
What is the order of the citric acid cycle beginning at succinate?
|
Sex Feels Marvelous Over Cordelia And If Kruti Sucks-a-Neil.
1. Succinate 2. Fumarate 3. Malate 4. Oxaloacetate 5. Citrate 6. cis-aconitate 7. Isocitrate 8. alpha-ketoglutarate 9. succinyl coA |
|
|
What is the order of the citric acid cycle beginning at fumarate?
|
1. fumarate
2. malate 3. oxaloacetate 4. citrate 5. cis-aconitate 6. isocitrate 7. alpha-ketoglutarate 8. succinyl coA 9. succinate |
|
|
What is the order of the citric acid cycle beginning at malate?
|
1. malate
2. oxaloacetate 3. citrate 4. cis-aconitate 5. isocitrate 6. alpha-ketoglutarate 7. succinyl coA 8. succinate 9. fumarate |
|
|
What is the order of the citric acid cycle beginning at oxaloacetate?
|
1. oxaloacetate
2. citrate 3. cis-aconitate 4. isocitrate 5. alpha-ketoglutarate 6. succinyl coA 7. succinate 8. fumarate 9. malate |
|
|
What stimulates and inhibits citrate synthase?
|
Stimulate: Nothing
Inhibit: ATP |
|
|
What stimulates and inhibits isocitrate dehydrogenase?
|
Stimulate: ADP
Inhibit: 1. ATP 2. NADH |
|
|
What stimulates and inhibits alpha-ketoglutarate dehydrogenase?
|
Stimulate: Nothing
Inhibit: 1. ATP 2. NADH 3. Succinyl CoA |
|
|
Which steps in the citric acid cycle produce CO2?
|
The steps where carbons are lost, the two structures after isocitrate each have one less carbon than the last.
1. Isocitrate to alpha-ketoglutarate 2. alpha-ketoglutarate to succinyl coA |
|
|
Which steps in the citric acid cycle produce reducing equivalents?
|
The only step that produces FADH2 is the only one that also yields an F product.
1. Isocitrate to alpha ketoglutarate (1 NADH) 2. alpha-ketoglutarate to succinyl coA (1 NADH) 3. Succinate to Fumarate (1 FADH2) 4. Malate to Oxaloacetate (1 NADH) |
|
|
Which steps in the citric acid cycle produce ATP?
|
None, however 1 GTP is produced from the conversion of Succinyl CoA to Succinate.
|
|
|
How much ATP is produced by the citric acid cycle per molecule of acetyl coA?
|
12 ATP.
3 NADH x 3 ATP/NADH= 9 ATP 1 FADH2 x 2 ATP/FADH2 = 2 ATP 1 GTP x 1 ATP/GTP = 1 ATP The total is 12 ATP |
|
|
How much ATP is produced by the citric acid cycle per molecule of glucose?
|
24
1 cycle: 3 ATP/NADH= 9 ATP 1 FADH2 x 2 ATP/FADH2 = 2 ATP 1 GTP x 1 ATP/GTP = 1 ATP The total is 12 ATP per acetyl coA. However, there are 2 acetyl coA molecules produced per glucose molecule. Thus the total is 24. |
|
|
Name the complexes and important coenzymes and cytochromes in the electron transport chain.
|
1. Complex I
2. Coenzyme Q 3. Complex III 4. Cytochrome C 5. Complex IV 6. Complex V |
|
|
Where in the electron transport chain do NADH and FADH2 release their electrons?
|
Complex I
|
|
|
Where in the electron transport chain is O2 reduced to 2H2O?
|
Complex IV
|
|
|
Where in the electron transport chain is ADP converted to ATP?
|
Complex V aka ATP synthase aka mitochondrial ATPase
|
|
|
Name three classes of oxidative phosphorylation poisons.
|
1. Electron transport inhibitors
2. ATPase inhibitors 3. Uncoupling agents |
|
|
What is the mechanism of electron transport inhibitors?
|
1. Directly inhibit electron transport causing:
2. Decreased protein gradient and decrease in O2 consumption, thereby: 3. Blocking ATP synthesis |
|
|
What is the mechanism of ATPase inhibitors?
|
1. Directly inhibit mitochondrial ATPase causing:
2. Increased protein gradient and increased oxygen consumption, but no ATP is produced because electron transport stops. |
|
|
What is the mechanism of uncoupling agents?
|
"Uncouples" ATP synthesis from gradient production
1. Increase permeability of membrane 2. Proton gradient decreases, but oxygen consumption increases, as the gradient is not being maintained. 3. ATP synthesis stops, but electron transport continues. |
|
|
What is rotenone?
|
An electron transport inhibitor.
|
|
|
What is the mechanism of CN?
|
Electron transport inhibition
|
|
|
What is the mechanism of CO?
|
Electron transport inhibition
|
|
|
What is antimycin A?
|
An electron transport inhibitor.
|
|
|
What is the mechanism of oligomycin?
|
ATPase inhibition
|
|
|
What is the mechanism of thermogenin?
|
Uncoupling protein OR UCP which is an uncoupling agent
|
|
|
Where is thermogenin found?
|
Brown adipose tissue
|
|
|
What is the mechanism of 2,4-dinitrophenol?
|
Uncoupling agent
|
|
|
Name three uncoupling agents
|
1. UCPs (such as Thermogenin)
2. 2,4-dinitrophenol 3. aspirin |
|
|
Name the irreversible enzymes in gluconeogenesis, and where they are found.
|
Pathway Produces Fresh Glucose
All the enzymes are found only in the liver, kidney, and intestinal epithelium 1. Pyruvate carboxylase in the mitochondria 2. PEP carboxykinase in the cytosol 3. Fructose-1,6-bisphosphatase in the cytosol 4. Glucose-6-Phosphatase in the endoplasmic reticulum |
|
|
Name the irreversible enzymes in glycolysis.
|
1. Hexokinase
2. Phosphofructokinase-1 3. Pyruvate kinase 4. Pyruvate dehydrogenase |
|
|
What are the requirements of PEP carboxykinase?
|
GTP
|
|
|
Where does the pentose phosphate pathway happen?
|
Cytoplasm of Red Blood Cells, and in lactating mammary glands, liver, and adrenal cortex (all sites of fatty acid or steroid synthesis except RBCs)
|
|
|
How much ATP is used in the pentose phosphate shunt?
|
None
|
|
|
What are the main products of the pentose phosphate shunt and their uses?
|
1. NADPH (for fatty acid and steroid synthesis, glutathione reduction, and cytochrome P-450)
2. Ribose-5-phosphate (for nucleotide synthesis) 3. G3P and F6P (glycolytic intermediates) |
|
|
What are the key enzymes of the pentose phosphate shunt and are the reactions reversible or irreversible?
|
1. Glucose-6-phosphate dehydrogenase (irreversible)
2. Transketolase (reversible) |
|
|
What does transketolase require?
|
Thiamine (Vitamin B1)
|
|
|
What is the rate-limiting enzyme in the Pentose phosphate pathway?
|
Glucose-6-Phosphate Dehydrogenase
|
|
|
What is glutathione used for?
|
Detoxification of free radicals and peroxides.
|
|
|
What does NADPH deficiency in RBCs result in?
|
Hemolytic anemia
|
|
|
Name some oxidizing agents that someone with a G6PD deficiency is vulnerable to.
|
1. Fava beans
2. Sulfonamides 3. Primaquine 4. Antituberculosis drugs |
|
|
What protection does G6PD deficiency provide?
|
Protection against malaria
|
|
|
Which group is more likely to have G6PD deficiency?
|
Blacks
|
|
|
What are Heinz bodies?
|
altered Hemoglobin precipitates within RBCs, found in G6PD deficiency
|
|
|
What histologic change is seen in G6PD deficiency
|
Heinz bodies within red blood cells
|
|
|
What is the etiology of fructose intolerance?
|
1. Lack of aldolase B
2. Build up of Fructose-1-Phosphate 3. Decrease in available phosphate 4. Inhibition of glycogenolysis and gluconeogenesis |
|
|
What is the clinical presentation of fructose intolerance?
|
hypoglycemia, jaundice, cirrhosis, and vomiting
|
|
|
What is the difference in presentation between von Gierke's disease and fructose intolerance?
|
Both have hypoglycemia, jaundice, cirrhosis and vomiting.
von Gierke's disease also has lactic acidosis whereas fructose intolerance does not. |
|
|
What is the treatment for fructose intolerance?
|
Decreased intake of both fructose and sucrose.
|
|
|
What is the etiology of essential fructosuria?
|
Defect in fructokinase leading to lack of metabolism of fructose. Benign and asymptomatic
|
|
|
What is the clinical presentation of essential fructosuria?
|
Fructose appears in the blood and urine
|
|
|
Which is more serious, essential fructosuria or fructose intolerance?
|
Fructose intolerance, because it depletes the cells of phosphate.
|
|
|
What is the etiology of classic galactosemia?
|
1. Absence of galactose-1-phosphate uridyl transferase
2. Build up of toxic substances including galactitol |
|
|
What is the presentation of classic galactosemia?
|
Early:
1. Galactosemia 2. Galactosuria 3. Vomiting 4. Diarrhea 5. Jaundice Late: 1. Cataracts 2. Hepatosplenomegaly 3. Mental retardation |
|
|
How does galactokinase deficiency present?
|
1. Galactosemia
2. Galactosuria More severe symptoms such as cataracts, hepatosplenomegaly and mental retardation can follow. |
|
|
What is the treatment for classic galactosemia?
|
Exclude galactose and lactose from the diet.
|
|
|
What enzyme converts galactose to galactitol?
|
Aldose reductase
|
|
|
What does aldose reductase do?
|
Converts galactose to galactitol
|
|
|
What enzyme converts Galactose to galactose-1-phosphate?
|
Galactokinase
|
|
|
What enzyme converts Galactose-1-Phosphate to Glucose-1-Phosphate?
|
Uridyl transferase
|
|
|
What enzyme converts UDP-galactose to UDP-glucose?
|
4-epimerase
|
|
|
What does galactokinase do?
|
converts Galactose to galactose-1-phosphate
|
|
|
What does 4-epimerase do?
|
converts between UDP-galactose and UDP-glucose
|
|
|
What does Uridyl transferase do?
|
1. converts UDP-glucose to UDP-galactose
2. converts Galactose-1-Phosphate to Glucose-1-Phosphate |
|
|
What enzyme converts UDP-glucose to UDP-galactose?
|
Uridyl transferase
|
|
|
Which groups are more likely to be lactose intolerant?
|
1. Blacks
2. Asians |
|
|
What is the etiology of lactose intolerance?
|
Loss of brush-border lactase
|
|
|
How does lactose intolerance present?
|
1. Bloating
2. Cramps 3. Osmotic diarrhea |
|
|
What is the treatment for lactose intolerance?
|
Avoid milk or add lactase pills to the diet
|
|
|
What are the essential amino acids?
|
PVT TIM HALL
1. Phenylalanine 2. Valine 3. Threonine 4. Tryptophan 5. Isoleucine 6. Methionine 7. Histidine 8. Alanine 9. Leucine 10. Lysine |
|
|
What are the conditionally essential amino acids, and why are they conditionally essential?
|
The condition is age. They are necessary early in life during growth.
Mnemonic: Babies CRY for Help 1. Cysteine 2. aRginine 3. tYrosine 4. Histidine |
|
|
Cysteine or Cystine: The amino acid
|
Cysteine
|
|
|
Cysteine or Cystine: Two copies of the amino acid joined by a disulfide bond
|
Cystine
|
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Phenylalanine
|
Essential
Both glucogenic and ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Valine
|
Essential
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Tryptophan
|
Essential
Both glucogenic and ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Threonine
|
Essential
Both glucogenic and ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Isoleucine
|
Essential
Both glucogenic and ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Methionine
|
Essential
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Histidine
|
Essential
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Arginine
|
Essential
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Leucine
|
Essential
Ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Lysine
|
Essential
Ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Tyrosine
|
Conditionally essential (during life and early growth)
(Phenylalanine and Tetrahydrobiopterin produce tyrosine and dihydrobiopterin) Both glucogenic and ketogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Glutamate
|
Inessential (made from alpha-ketoglutarate)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Aspartate
|
Inessential (made from asparagine or oxaloacetate by aspartate aminotransferase)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Proline
|
Inessential (Glutamate makes proline and ornithine)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Glycine
|
Inessential (synthesized during reactions involving tetrahydrofolate)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Cysteine
|
Conditionally essential (during life and early growth)
(Methionine begets S-adenosyl methionine which begets intermediates which beget cysteine) Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Alanine
|
Inessential (made from pyruvate by alanine aminotransferase in the Cori cycle)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Serine
|
Inessential (made from a descendant of 3PG and with an amine group from glutamate)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Glutamine
|
Inessential (made from glutamate)
Glucogenic |
|
|
Is the following amino acid essential or inessential, and is it glucogenic, ketogenic, or both?: Asparagine
|
Inessential (made from aspartate)
Glucogenic |
|
|
Which amino acids are acidic?
|
Aspartate and glutamate are negatively charged at body pH
|
|
|
Which amino acids are basic?
|
Arginine, Lysine and Histidine
Arginine and Lysine are increased in histones which bind negatively charged DNA. Histidine has no charge at body pH. |
|
|
Zinc deficiency: Presentation
|
"Delayed wound healing, hypogonadism, and decreased adult hair (axillary, facial, pubic)"
|
|
|
Zinc deficiency: Predisposes to what?
|
Alcoholic cirrhosis
|
|
|
Ethanol metabolism: All steps with enzymes and cofactors
|
"Step 1: Ethanol is oxidized by NAD (forming NADH) to acetaldehyde using alcohol dehydrogenase. Step 2: Acetaldehyde is oxidized by NAD (forming NADH) to acetate using acetaldehyde dehydrogenase."
|
|
|
Ethanol metabolism: Limiting reagent
|
NAD+
|
|
|
Ethanol metabolism: Order of kinetics of alcohol dehydrogenase
|
Zero-order kinetics
|
|
|
Disulfiram: Mechanism
|
"Disulfiram inhibits acetaldehyde dehydrogenase, leading to an accumulation of acetaldehyde, leading to increased hangover symptoms."
|
|
|
Which drug inhibits acetaldehyde dehydrogenase?
|
Disulfiram
|
|
|
Ethanol hypoglycemia: mechanism
|
"1. Ethanol metabolism increases NADH/NAD ratio in the liver. 2. Pyruvate and oxaloacetate are reduced by NADH respectively to lactate and malate. 3. Decreased pyruvate and oxaloacetate leads to decreased gluconeogenesis. 4. Decreased gluconeogenesis leads to hypoglycemia."
|
|
|
What are the consequences of the altered NADH/NAD ratio seen in alcoholics?
|
"Short-term: Hypoglycemia, Long-term: Hepatic fatty change"
|
|
|
What is the mechanism behind chronic fatty change in alcoholics?
|
"1. Ethanol metabolism leads to an increased NADH/NAD ratio in the liver. 2. This ratio prefers fatty acid synthesis over glycolysis."
|
|
|
Kwashiorkor: Clinical picture
|
Small child with a swollen belly and depigmented hair.
|
|
|
Kwashiorkor: Clinical presentation
|
"Kwashiorkor results from protein-deficient MEALS. Malabsorbtion, Edema, Anemia, Liver (fatty change), Skin lesions"
|
|
|
Protein malnutrition leads to what disease?
|
Kwashiorkor (as opposed to Marasmus from energy malnutrition)
|
|
|
Energy malnutrition leads to what disease?
|
Marasmus (as opposed to Kwashiorkor from protein malnutrition)
|
|
|
Marasmus: Clinical presentation
|
"Tissue and muscle wasting, loss of subcutaneous fat, and variable edema"
|
|
|
"Chromatin structure: In the beads on a string analogy, what are the beads?"
|
"Start with a nucleosome core made up of an 8 histone cube (two each of positively-charged histones H2A, H2B, H3, and H4). Negatively charged DNA loops twice around nucleosome core."
|
|
|
"Chromatin structure: In the beads on a string analogy, what is the string and how long is it?"
|
Histone H1 ties the nucleosomes together in a 30-nm fiber string
|
|
|
Chromatin structure: What histones are included and which of these are not in the nucleosome core?
|
"H1 (only one not in the core), H2A, H2B, H3, and H4"
|
|
|
Heterochromatin or Euchromatin: Which is more condensed?
|
Heterochromatin. Euchromatin is less condensed.
|
|
|
Heterochromatin or Euchromatin: Which is less condensed?
|
Euchromatin. Heterochromatin is more condensed.
|
|
|
Heterochromatin or Euchromatin: Which is transcriptionally active?
|
"Euchromatin (""eu"" means true, so think ""truly transcribed"")"
|
|
|
Heterochromatin or Euchromatin: Which is transcriptionally inactive?
|
Heterochromatin
|
|
|
Name the purines.
|
Adenine and Guanine
|
|
|
Name the pyrimidines.
|
"Cytosine, Uracil, Thymine"
|
|
|
Which base pair bond has 3 Hydrogen bonds?
|
Guanine to Cytosine
|
|
|
Which base pair bond has 2 Hydrogen bonds?
|
Adenine to Thymine
|
|
|
How many Hydrogen bonds does the Guanine to Cytosine pairing have?
|
3
|
|
|
How many Hydrogen bonds does the Adenine to Thymine pairing have?
|
2
|
|
|
Which amino acids are necessary for purine synthesis?
|
"Glycine, Aspartate, Glutamine"
|
|
|
"In nucleic acids, what kind of substitution is a transition?"
|
"TransItion = Identical type (Purine for purine or pyrimidine for pyrimidine")
|
|
|
"In nucleic acids, what kind of substitution is a transversion?"
|
"TransVersion = conVersion between types (Purine for pyrimidine or vice versa")
|
|
|
What does it mean for genetic code to be unambiguous?
|
Each codon specifies only one amino acid.
|
|
|
What does it mean for genetic code to be degenerate?
|
More than one codon may code for the same amino acid.
|
|
|
What does it mean for genetic code to be redundant?
|
More than one codon may code for the same amino acid.
|
|
|
Which amino acid is coded by only one codon?
|
Methionine
|
|
|
"~ average pKa of carboxyl group on AA"
|
2.3
|
|
|
"~ pKa of side chain of Aspartic Acid"
|
"<4"
|
|
|
"~ pKa of side chain of Glutamic Acid"
|
">4"
|
|
|
"~ pKa of side chain of Histidine"
|
6
|
|
|
"~ pKa of side chain of Cysteine"
|
8
|
|
|
"~ average pKa of amino group on AA"
|
9.6
|
|
|
"~ pKa of side chain of Tyrosine"
|
10
|
|
|
"~ pKa of side chain of Lysine"
|
10.5
|
|
|
"~ pKa of side chain of Arginine"
|
12.5
|
|
|
"An acid with a pKa of x serves as a buffer best at x + what?"
|
"positive or negative 1 (equal amounts of charged and uncharged acid)"
|
|
|
"Trypsin cleaves peptides at which side of what residues?"
|
"C-terminal of lysine or arginine (the most basic amino acids)"
|
|
|
"Cyanogen bromide cleaves peptides at which side of what residues?"
|
"C-terminal of methionine"
|
|
|
"Pepsin cleaves peptides at which side of what residues?"
|
"C-terminal side of tyrosine, phenylalanine, and tryptophan (all have phenyl groups, these are the same bonds as chymotrypsin. Pepsin's action ceases when the NaHCO3 raises the pH of the intestinal contents)"
|
|
|
"Chymotrypsin cleaves peptides at which side of what residues?"
|
"C-terminal side of tyrosine, phenylalanine, and tryptophan residues (all have phenyl groups, these are the same bonds as pepsin, whose action ceases when the NaHCO3 raises the pH of the intestinal contents)."
|
|
|
"# of aas in one turn of alpha-helix"
|
3.6
|
|
|
"Amino acids that disrupt alpha-helix"
|
"proline, many charged aas, bulky side chains"
|
|
|
"Which reagent sequentially removes N-terminal residues from a polypeptide?"
|
"Phenylisothiocyanate (Edman degradation)"
|
|
|
"Which reagent sequentially removes C-terminal residues from a polypeptide?"
|
"Carboxypeptidase"
|
|
|
"What kind of inheritance and mutation is the alpha-1-antitrypsin deficiency?"
|
"Autosomal recessive, single purine substitution (GAG to AAG)"
|
|
|
"Anode: What does it attract?"
|
"Anions"
|
|
|
"Anode: What does it contain?"
|
"Cations"
|
|
|
"Cathode: What does it attract?"
|
"Cations"
|
|
|
"Cathode: What does it contain?"
|
"Anions"
|
|
|
"Inhibitors of electron transport from FMNH2 to Coenzyme Q"
|
"Amytal and Rotenone"
|
|
|
"Inhibitors of electron transport from Cytochrome b to Cytochrome c"
|
"Antimycin A"
|
|
|
"Inhibitors of electron transport from Cytochrome a+a3 to Oxygen"
|
"Cyanide, CO, and Sodium azide"
|
|
|
"Where do GLUT1 receptors predominate over other GLUT receptors?"
|
"RBCs"
|
|
|
"Where do GLUT4 receptors predominate over other GLUT receptors?"
|
"Adipose tissue and skeletal muscle"
|
|
|
"Which tissues have cotransport of glucose?"
|
"Epithelial cells of the intestine, renal tubular cells, and choroid plexus"
|
|
|
"Which tissues (7) need glucose as fuel?"
|
"Brain, RBCs, Renal medulla, lens, cornea, testes, exercising muscle"
|
|
|
"Where is pyruvate carboxylase found and not found?"
|
"Found in mitochondria of liver and kidney cells, not foudn in mitochondria of muscle"
|
|
|
"Where is Fructose 1-6 bisphosphatase found?"
|
"Liver and kidney"
|
|
|
"What is the Cori cycle?"
|
"Lactate in muscle is shuttled to liver where it is turned into glucose."
|
|
|
"How does glucagon stimulate gluconeogenesis?"
|
"Regulation of F2,6-BP and inactivation of Pyruvate Kinase via elevation of cAMP-dependent protein kinase A."
|
|
|
"This oxidation accounts for about two thirds of the total oxygen consumption and ATP production in most animals, including humans."
|
"Oxidation of acetyl coA to CO2 and H2O."
|
|
|
"What inhibits pyruvate dehydrogenase?"
|
"Acetyl CoA and NADH (no need for more of either). These activate PD kinase (Phosphorylates enzyme with ATP, which must be in abundance, so no more is needed)"
|
|
|
"What stimulates pyruvate dehydrogenase?"
|
"ADP (need more ATP. Inhibits PD kinase and stimulates PD phosphatase.)"
|
|
|
"Which is active?: Phosphorylated or dephosphorylated pyruvate dehydrogenase"
|
"Dephosphorylated."
|
|
|
"What inhibits citrate synthase?"
|
"ATP and NADH (no need for more of either), Succinyl CoA (""Slow down partner, the guys ahead of you are trying to do their job!""), Acyl CoA fatty acid derivatives (Citrate provides acetyl CoA to synthesize fatty acids and activates acetyl CoA carboxylase, rate limiting enzyme of fatty acid synthesis)."
|
|
|
"Where in glycolysis and TCA does CO2 come off?"
|
"3 places: Pyruvate to Acetyl CoA, Isocitrate to alpha-ketoglutarate, and alpha-ketoglutarate to Succinyl CoA"
|
|
|
"What is the rate-limiting step of the TCA?"
|
"Isocitrate to alpha-ketoglutarate by isocitrate dehydrogenase"
|
|
|
"What activates isocitrate dehydrogenase?"
|
"ADP"
|
|
|
"What inhibits isocitrate dehydrogenase?"
|
"ATP and NADH"
|
|
|
"Sources of Succinyl CoA"
|
"TCA intermediate, and from odd chained fatty acids, and from propionyl coA from metabolism of branched-chain amino acids."
|
|
|
"Uses of Succinyl CoA"
|
"TCA intermediate, and biosynthesis of heme"
|
|
|
"Where in the TCA does NADH come from?"
|
"Pyruvate to Acetyl CoA, Isocitrate to alpha-ketoglutarate, alpha-ketoglutarate to succinyl coA, Malate to Oxaloacetate"
|
|
|
"Where in the TCA does FADH2 come from my dear?"
|
"Succinate to fumarate my sweet."
|
|
|
"Why is FAD used to oxidize succinate?"
|
"Succinate is not powerful enough to reduce NAD."
|
|
|
"What are the important products of the HMP pathway?"
|
"2 NADPH, Ribose, and glyceraldehyde-3-Phosphate and Fructose-6-phosphate"
|
|
|
"Which major metabolic reactions require Thiamine as a cofactor?"
|
"TCA: Pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase, HMP shunt: Transketolase"
|
|
|
"What is NADPH used for?"
|
"1. Reductive biosynthesis (eg fatty acids and steroids) 2. Reduction of oxygen directly (myeloperoxidase system's famed respiratory burst) and hydrogen peroxide indirectly (through reduction of glutathione) 3. Cytochrome P-450 mono-oxygenase system"
|
|
|
"What is the famed respiratory burst?"
|
"The rapid conversion of O2 to superoxide using NADPH."
|
|
|
"What disease process is due to a missing respiratory burst?"
|
"Chronic granulomatous disease"
|
|
|
"Where is the mutation for G6PD?"
|
"Point mutation in coding region of the G6PD gene (X-linked)"
|
|
|
"What is the relation of polyols to sugars?"
|
"Polyols are monosaccharides where the carbonyl group is reduced to an alcohol."
|
|
|
"What is a glycoside?"
|
"Carbohydrate attached to non-carbohydrate structures."
|
|
|
"What is a reducing sugar?"
|
"A monosaccharide where the anomeric carbon (Carbon 1) is free."
|
|
|
"What is the result of lack of disaccharidase activity of intestinal mucosa?"
|
"Osmotically active disaccharides suck water out of mucosa causing osmotic diarrhea."
|
|
|
"Where is fructokinase found?"
|
"Liver (processes most dietary fructose), kidney, small intestine"
|
|
|
"Why is fructose metabolism faster than glucose metabolism?"
|
"Bypasses PFK, major regulatory step of glycolysis."
|
|
|
"What enzyme is missing in hereditary fructose intolerance?"
|
"Aldolase B"
|
|
|
"What does aldose reductase do?"
|
"Reduces glucose to sorbitol"
|
|
|
"Where is aldose reductase found?"
|
"Lens, retina, Schwann cells, kidney, placenta, RBCs, and gonads"
|
|
|
"What does sorbitol dehydrogenase do?"
|
"Oxidizes sorbitol to fructose."
|
|
|
"Where is sorbitol dehydrogenase found?"
|
"Liver and gonads (ovaries, seminal vesicles, sperm)"
|
|
|
"Mechanism of sorbitol toxicity"
|
"Extra glucose freely enters cells containing aldose reductase which converts it to sorbitol. Sorbitol may not pass through, and low or absent sorbitol dehydrogenase prevents it from being changed to fructose. Strong osmotic effects lead to swelling and damage."
|
|
|
"Chondroitin Sulfate: Where found?/Distinguishing characteristic from other GAGs"
|
"Cartilage, tendons, ligaments, aorta. Most abundant GAG in body."
|
|
|
"Chondroitin Sulfate: Use/Mechanism"
|
"Form proteoglycan aggregates. Cartilage: Bind collagen and hold fibers in a tight, strong network"
|
|
|
"Dermatan Sulfate: Where found?/Distinguishing characteristic from other GAGs"
|
"Found in skin, blood vessels, and heart valves"
|
|
|
"Dermatan Sulfate: Use/Mechanism"
|
|
|
|
"Keratan Sulfate: Where found?/Distinguishing characteristic from other GAGs"
|
"Found in cartilage proteoglycan aggregates with chondroitin sulfate, and in cornea. Most heterogeneous GAG."
|
|
|
"Keratan Sulfate: Use/Mechanism"
|
|
|
|
"Heparin: Where found?/Distinguishing characteristic from other GAGs"
|
"Intracellular compound (unlike other GAGs). Found in mast cells of artery walls, especially in lungs, liver, and skin"
|
|
|
"Heparin: Use/Mechanism"
|
"Anticoagulant"
|
|
|
"Heparan Sulfate: Where found?/Distinguishing characteristic from other GAGs"
|
"Extracellular, unlike heparin. Found in basement membrane and as a ubiquitous component of cell surfaces."
|
|
|
"Heparan Sulfate: Use/Mechanism"
|
|
|
|
"Hyaluronic Acid: Where found?/Distinguishing characteristic from other GAGs"
|
"Found in synovial fluid of joints, vitreous humor f eye, umbilical cord, and loose connective tissue. Unlike other GAGs: Unsulfated, not covalently attached to protein, and only GAG not limited to animal tissue, but also found in bacteria."
|
|
|
"Hyaluronic Acid: Use/Mechanism"
|
"Lubricant and shock absorber"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Enzyme deficiency"
|
"Hunter's: Iduronate sulfatase, Hurler's: alpha-L-iduronidase"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Corneal clouding?"
|
"Hunter's: No, Hurler's: Yes"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Mental retardation?"
|
"Both (Hunter's ranges from mild to severe)"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Physical deformity?"
|
"Hunter's: Mild to severe, Hurler's: Dwarfing, coarse facial features, (gargoylism)"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Which GAGs' degradation is affected?"
|
"Both: Dermatan sulfate and Heparan sulfate"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Severity?"
|
"Hunter's: Less Hurler's: More"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Inheritance?"
|
"Hunter's: X-linked Recessive, Hurler's (and all other mucopolysaccharidoses): Autosomal recessive"
|
|
|
"Hunter's Syndrome vs Hurler's Syndrome: Aggressive behavior?"
|
"Hunter's: Yes, Hurler's: No"
|
|
|
"Mnemonic for Hurler's syndrome: HURLERS. What does it stand for?"
|
"H: Hepatosplenomegaly/Heparan and Dermatan sulfate, U:Ugly facies, R: aRteries filled with GAGs, L: L-iduronidase, E: Eyes clouded, early death, R: Retardation/Respiratory obstruction, S: Short/stubby fingers"
|
|
|
"I-Cell disease: Pathophysiology"
|
"Inability of cell to phosphorylate mannose residues on glycoproteins indicating that they are lysosome bound."
|
|
|
"I-Cell disease: Presentation"
|
"Skeletal abnormalities, restricted joint movement, coarse facial features, severe psychomotor impairment, death by 8 years"
|
|
|
"Refsum Disease: Pathophysiology"
|
"Inability to degrade phytanic acid, resulting in accumulation in plasma and tissues"
|