Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
26 Cards in this Set
- Front
- Back
|
C= Coloboma
H= Heart Defect A= Atresia choanae R= retardation of growht and development G= Genital Anomalies E= Ears |
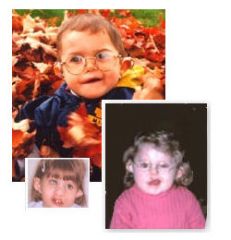
What is this syndrome?
|
|
|
Iris Coloboma, a defect or incomplete congenital development of the iris.
|
What does this picture depict?
|
|
|
What are the characteristics of Angelman's Syndrome?
|
Somtimes this syndrome is called the "happy puppet" syndrome. Affected children are normally grown and normally formed at birth. They experience global developmental delay, but speech is the most affected area, and most children never speak. They laugh frequently and have an atxic gait, often holding their elbows away from their bodies as they walk.
|
|
|
How do you make the diagnosis of Marfan syndrome?
|
To meet the diagnostic criteria for Marfan syndrome, an individual must have a major criterion in two separate systems and involvement of a third system. If the individual has a confirmed positive family history for Marfan syndrome in a first degree relative, he or she must have a major criterion in one system and involvement of another system to meet the criteria for diagnosis. The systems are: skeletal, pulmonary, cardiovascular, ocular and integumentary.
|
|
|
What are the major criteria for Marfan syndrome?
|
at least four major skeletal features, aortic root dilation, subluxation of the lens of the ey, and dural ectasia which is idenitified on MRI or CT of lumbar spine.
|
|
|
How do you diagnose Neurofibromatosis 1?
|
Two or more of the following:
1. six or more cafe au lait macules measuring greater than 0.5 cm in diameter in prepubertal individuals and greater than 1.5 cm in those who are postpubertal. 2. More than 2 neurofibromas of any type or more than one plexiform neurofibroma 3. Freckling in the axillary and or inguinal regions 4. Optic Glioma 5. More than 2 lisch nodules or iris hamartomas 6. dysplasia of the sphenoid bone or dysplasia of thinning of long bone cortex. 7. a first degree relative who has NF-1 |
|
|
What are the frequencies of freckling?
|
Axillary or inguinal freckling occurs in 85% of patients who have NF-1 by age 10 years.
|
|
|
What are the frequencies of lisch nodules?
|
Lisch nodules are iris hamartomas that are observed best by slitlamp exam and are present in 25% of those who have NF-1 by age 5 years, 50% by age 10 years and more than 95% by age 20 years.
|
|
|
What are the frequencies of neurofibromas?
|
Neurofibromas do not develp until late child hood or adolescence; they are present in 20% of affected individuals by age 10 years and in more than 90% of adults.
|
|
|
Congenital syphilis
Acromegaly Russell-Silver syndrome (Russell-Silver dwarf) Pfeiffer syndrome Basal cell nevus syndrome Crouzon syndrome Hurler syndrome Rubinstein-Taybi syndrome Fetal trimethadione syndrome Cleidocranial dysostosis |
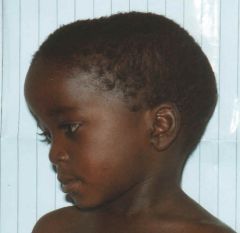
What does this child have and what are the associated syndromes?
|
|
|
What syndromes are associated with Craniosynostosis?
|
Apert and Crouzon.
|
|
|
What is Apert syndrome?
|
Apert's syndrome (acrocephalosyndactyly) is an autosomal dominant disorder that occurs in one of every 160,000 live births.23 The syndrome is caused by nucleotide alterations resulting in amino-acid substitutions, leading to a mutation in the FGFR2 gene located on chromosome 10. Craniosynostosis and symmetric syndactyly of the extremities are hallmarks of this syndrome. The clinical features include misshapen skull caused by coronal suture synostosis, wide-set eyes, midface hypoplasia, choanal stenosis, and shallow orbits. Intracranial anomalies include megalocephaly, hypoplastic white matter, and agenesis of the corpus callosum, leading to cognitive impairment. Cardiac anomalies, including atrial septal defect and ventricular septal defect, and renal anomalies such as hydronephrosis occur in 10 percent of these patients.
|
|
|
What is Crouzon syndrome?
|
Crouzon's disease is inherited through an autosomal-dominant pattern.23 Nearly 60 percent of cases are new mutations, and many are associated with paternal age older than 35 years. Crouzon's disease occurs in one of every 25,000 live births and accounts for 5 percent of cases of craniosynostosis. Nucleo-tide alterations causing amino-acid substitutions at the FGFR2 gene on chromosome 10 lead to the Crouzon phenotype. Clinical findings include brachycephalic craniosynostosis, significant hypertelorism, proptosis, maxillary hypoplasia, beaked nose and, possibly, cleft palate. Intracranial anomalies include hydrocephalus, Chiari 1 malformation, and hindbrain herniation (70 percent). Pathology of the ear and cervical spine is common. Infants with Crouzon's disease do not have anomalies of the hands and feet as do infants with Apert's syndrome.
|
|
|
What tests should ordered for craniosynostosis?
|
Perform skull radiography with anterior-posterior, lateral, and Water views. Prematurely fused sutures are easily identified by the absence of sutures and associated ridging of the suture line. Sutures either are not visible or have evidence of sclerosis.
Cranial CT scan with 3-dimensional reconstruction is not required in most infants. It is sometimes performed when surgery is being considered. Nuclear medicine isotope studies are of limited value. Other Tests: Endocrine evaluation: Order thyroid and parathyroid studies when associated features suggest these diagnoses. |
|
|
What is choanal atresia associated with?
|
Bilateral choanal atresia is commonly associated with other congenital anomalies. The CHARGE association (Coloboma, Heart defects, choanal Atresia, Retarded growth, Genitourinary abnormalities, and Ear anomalies) is present to varying extent in approximately 50% of bilateral cases. Bilateral choanal atresia has been reported in association with other craniofacial syndromes and skull-based defects including encephalocele. Most unilateral cases are isolated anomalies.
|
|
|
CHOANAL ATRESIA IS a congenital anomaly of the anterior skull base characterized by closure of one or both posterior nasal cavities. The condition occurs in 1 out of every 7,000 to 8,000 live births. Approximately 60% of reported cases are unilateral with a right-sided predominance.
|
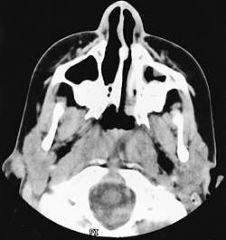
What does the picture depict?
|
|
|
Dandy-Walker malformation is a rare congenital malformation and involves the cerebellum and fourth ventricle. The condition is characterized by agenesis or hypoplasia of the cerebellar vermis, cystic dilatation of the fourth ventricle, and enlargement of the posterior fossa. A large number of concomitant problems may be present, but the syndrome exists whenever these 3 features are found. Approximately 70-90% of patients have hydrocephalus, which often develops postnatally. Dandy-Walker malformation may be associated with atresia of the foramen of Magendie and, possibly, the foramen of Luschka.
|
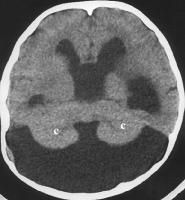
What does this picture depict?
|
|
|
Fetal Alchohol Syndrome
|
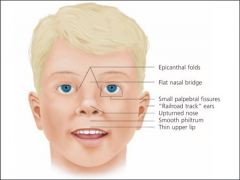
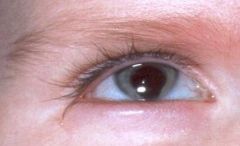
What does this picture depict?
|
|
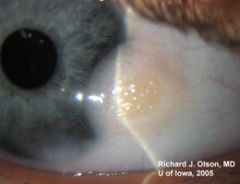
What else is associated with this eye finding?
|
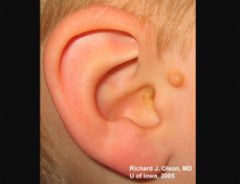
Goldenhar syndrome (also known as oculo-auriculo-vertebral spectrum or hemifacial microsmia) is a developmental malformation of the first and second branchial arches. --epibulbar dermoids, auricular appendages, malformations of the auricle, and hemifacial microsomia. Given the heterogeneous and variable presentation of this condition, it is probably best to describe it as a spectrum of dysmorphogenesis, the main characteristics of which are 1) epibulbar dermoids, 2) auricular appendages or malformations and 3) skeletal anomalies (including hemifacial microsomia and/or hemivertebra or butterfly vertebra. Other associated facial defects can include microphthalmos, strabismus, eyelid coloboma, preauricular fistulas, macrostomia, maxillary and mandibular hypoplasia, cleft palate, cleft lip, and middle and/or inner ear deformities contributing to hearing loss. In the majority of cases, only one side of the body is affected (interestingly, the right side is usually the more severely affected). Some affected individuals may have congenital heart defects, renal defects (hypoplasia or agenesis), or central nervous sytem (CNS) malformations (intracranial lipomas, hydrocephalus, cranial nerve dysgenesis and mental retardation). No formal minimal diagnostic criteria have been widely accepted, though some have been suggested . No inheritance pattern.
|
|
|
Treacher Collins.
Autosomal Dominant Sounds like Goldenhar Syndrome |
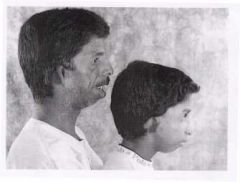
Most affected individuals have underdeveloped facial bones, particularly the cheek bones, and a very small jaw and chin (micrognathia). Some people with this condition are also born with an opening in the roof of the mouth called a cleft palate. In severe cases, underdevelopment of the facial bones may restrict an affected infant's airway, causing potentially life-threatening respiratory problems.
Some have coloboma. Some affected individuals have additional eye abnormalities that can lead to vision loss. This condition is also characterized by absent, small, or unusually formed ears. Defects in the middle ear (which contains three small bones that transmit sound) cause hearing loss in about half of cases What is it? Transmission? Similar to? |
|
|
Treacher Collins has features similar to what other syndromes?
|
Features of Treacher Collins syndrome (TCS) are also associated with Goldenhar syndrome, Nager syndrome, Miller syndrome, Pierre Robin sequence, and nonsyndromic mandibular hypoplasia
|
|
|
Which syndromes have no mental retardation attached?
|
Goldenhar Syndrome which is sporadic, Treache Collins which is autsomal dominant, VATERS.
|
|
|
Poland anomaly (PA) is described as an underdevelopment or absence of the chest muscle (pectoralis) on one side of the body and webbing of the fingers (cutaneous syndactyly) of the hand on the same side (ipsilateral hand). Sometimes referred to as "Poland syndrome," it is an uncommon condition present at birth (congenital). For people born with PA, the breastbone portion (sternal) of the pectoralis is also missing.
|
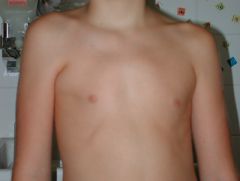
What is this syndrome?
|
|
|
Is Poland syndrome common in boys or girls? what is the etiology? Other features?
|
Common in males and has unknown etiology. 75% on right side. May have hypoplasia distally with syndactyly, brachydactyly and or oligodactyly.
|
|
|
You have attended to this child's emergent issues, and the seizures have stopped. You and the attending now have time to perform a more thorough physical exam. You've recently found that you are reading Smith's Recognizable Patterns of Human Malformation on a weekly basis, and have started to develop an eye for dysmorphic features. You expertly point out to your attending that this child is quite small for his age as well as microcephalic. In addition, you notice that he has low-set ears, ptosis, and epicanthal folds. His face is also notable for anteverted nostrils and a cleft palate. He has syndactyly of his 2nd and 3rd toes and single palmar creases on both hands. His parents give a history of severe developmental delay as well as a long history of failure to thrive. What makes the diagnosis for you is his genital exam: he has hypospadias and a bifid scrotum. What is the diagnosis and defect?
|
This is Smith Lemli Opitz Syndrome, a defect in cholesterol synthesis.
|
|
|
How do you diangose Smith Lemli Opitz?
|
Low plasma cholesterol, elevated plasma 7-dehydrochoesterol (precursor
|