Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
213 Cards in this Set
- Front
- Back
|
Anemia- general characteristics |
1. a reduction in Hct or Hgb concentration
2. When red cell mass (as measure by Hgb or less precisely by Hct) decreases, several compensatory mechanism maintain oxygen delivery to the tissues - Increased cardiac output (HR and SV) - increased extraction ratio - rightward shift of oxyhemoglobin curve (increases 2,3, diphosphoglycerate - 2,3 DPG) - expansion of plasma volume 3. when anemia develops rapidly, symptoms are more likely to be present, because there is little time for compensatory mechanisms. when onset is gradual, compensatory mechanisms are able to maintain oxygen delivery, and symptoms may be minimal or absent |
|
|
When is blood transfusion recommended?
|
- The hemoglobin concentration is < 7 g/dL OR
- the patient requires increased oxygen-carrying capacity (e.g. patients with CAD or some other cardiopulmonary disease) |
|
|
Clinical features of anemia
|
1. A variety of non-specific complaints- headache, fatigue, poor concentration, diarrhea, nausea, vague abdominal discomfort
2. Pallor - best noted in the conjunctive 3. hypotension and tachycardia 4. signs of the underlying cause- jaundice if hemolytic anemia, blood in the stool if GI bleeding |
|
|
diagnosis of anemia
|
1. Hb and Hct
- formula for converting Hgb to Hct : Hgb x 3 = Hct (1 units of PRBCs increases Hgb by 1 point and Hct by 3 points) 2. Reticulocyte index- the reticulocyte index is an important initial test in evaluating anemia because it indicates whether effective erythropoesis is occurring in the bone marrow. Effective erythrpoeisis is dependent on adequate raw materials (iron, B12, folate) in the bone marrow, absense of instrinsic bone marrow disease (e.g. aplastic anemia), adequate EPO from the kidney, and survival of reticuloctyes (no premature destruction before leaving the bone marrow) - a retic index > 2% implies excessive RBC destruction or blood loss. The bone marrow is responding to increased RBC requirements - a retic index < 2% implies inadequate RBC production by the bone marrow. 3. blood smear and RBC indices (especially Mean Corpuscular Volume - MCV) |
|
|
How does 1 unit of blood increase Hgb and Hct?
|
- formula for converting Hgb to Hct : Hgb x 3 = Hct
- 1 unit of PRBCs increases Hgb by 1 point and Hct by 3 points |
|
|
Diagnosing the cause of anemia- a general approach
|
1. If reticulocyte index < 2% then examine blood smear and RBC indices such as MCV
- MCV < 80 -- microcytic anemia - MCV > 100 -- macrocytic anemia - normocytic anemia * see other cards for potential causes of these 3 types of anemia |
|
|
causes of microcytic anemias
|
TAILS- Thalassemia, Anemia of Chronic Disease, Iron Deficiency, Lead Poisoning and Sideroblastosis
- if microcytic anemia ( MCV < 80) the differential includes: a. iron deficiency anemia- most common cause b. anemia of chronic disease - iron is present in the body by not available for hgb synthesis as it is trapped in macrophages c. thalassemias- defective synthesis of globin chains d. ring sideroblastic anemias (lead poisoning, pyridoxine deficiency, toxic effects of alcohol) - this is defective synthesis of protoporphoryins. Iron accumulates in the mitochondria |
|
|
causes of macrocytic anemias (MCV >100)
|
Differential includes
- nuclear defect (MCV increases significantly) - vitamin B12 and folate deficiencies - liver disease (MCV up to 115) 0 due to altered metabolism of plasma lipoproteins into their membranes, altering RBC shape (and increasing volume) - stimulated erythropoesis (MCV up to 110)- reticulocytes are larger than mature RBCs, resulting in and increase polychromatophylic RBCs |
|
|
Causes of normocytic anemia
|
- aplastic anemia
- bone marrow fibrosis - tumor - anemia of chronic disease (chronic inflammation, malignancy) - renal failure (dec EPO production) |
|
|
What should you do in anemia with a reticulocyte index > 2%?
|
suspect blood loss and look for the source of bleeding or suspect hemolysis (order bili, alk phos)
|
|
|
What is the most common class of anemias worldwide?
|
Microcytic anemias (MCV < 80)
|
|
|
What is most common cause of iron deficiency anemia in adults?
|
chronic blood loss.
- menstrual blood loss is the most common source. - in the absence of menstrual bleeding, GI blood loss is the most likely |
|
|
What age groups are most commonly affected by a dietary deficiency of iron?
|
1. infants and toddlers- occurs especially if the diet is predominantly human milk, which is low in iron. Children in this age group also have increased requirement for iron because of accelerate growth. It is most common between 6 mo and 3 years of age
2. Adolescents - rapid growth increases iron requirements. Adolescent women are at a particularly high risk due to menstrual blood loss 3. Pregnant women- increased demand |
|
|
Clinical features or iron deficiency anemia (IDA)
|
1. Pallor
2. Fatigue, generalized weakness 3. Dyspnea on exertion 4. Orthostatic lightheadedness 5. hypotension, if acute blood loss 6. Tachycardia, compensatory |
|
|
Diagnosis of iron deficiency anemia
|
1. Lab tests - decreased serum ferritin (most reliable test available), increased TIBC/transferrin levels, low TIBC saturation, decreased serum iron, microcytic, hyprochromic RBCs on peripheral smear.
2. bone marrow biopsy- the gold standard, but rarely performed. Indicated if laboratory evidence of IDA is present and no source of blood loss is found 3. If GI bleeding is suspected-- guaiac stool test or colonoscopy. Colon cancer is the most common cause of GI bleeding in the elderly |
|
|
What is the most common cause of GI bleeding in the elderly?
|
colon cancer
|
|
|
Treatment of Iron Deficiency Anemia (IDA)
|
1. Oral iron replacement (ferrous sulfate)
- a trial should be given to any menstruating woman. However, in men and postmenopausal women, there should be an attempt to determine the source of the blood loss - Side effects include constipation, nausea and dyspepsia 2. Parenteral iron replacement - Iron dextran can be administered IV or IM - This is rarely necessary because most patients respond to oral iron therapy. It may be useful in patients with poor absorption, patients that require more iron that oral therapy can provide or patients who cannot tolerate oral ferrous sulfate 3. Blood transfusion is not recommended unless anemia is severe or the patient has cardiopulmonary disease |
|
|
Thalassemias- general characteristics
|
1. inherited disorders characterized by inadequate production of either alpha or beta-globin chain of hemoglobin. Disorder is characterized according to which chain is deficient
2. Beta-thalassemias-- beta chain of hemoglobin is deficient, but the synthesis of alpha chains is unaffected. excess alpha chains bind to and damage the RBC membrane. It is most often found in people of Mediterranean, Middle Eastern, and Indian ancestry 2. Alpha thalassemias- There is a decrease in alpha chains, which are a component of all types of hemoglobin, The beta globin chains form tetramers, which are abnormal hemoglobins. The severity depends on the number of the gene loci that are deleted/mutated - it ranges from an asymptomatic carrier state to prenatal death |
|
|
Beta-thalassemia major
|
- Aka Cooley's anemia, homozygous B-chain thalassemia- occurs predominantly in Mediterranean populations
- Clinical features: severe anemia (microcytic hypochromic), massive hepatosplenomegaly, expansion of the bone marrow space which can cause distortion of bones, growth retardation and failure to thrive. If untreated (with blood transfusions) death occurs within the first few years of life secondary to progressive CHF - skull x-ray may show "crew-cut" appearance |
|
|
Diagnosis of beta-thalassemia
|
Hemoglobin electrophoresis - Hb F and Hb A2 are elevated
- peripheral blood smear shows a microcytic hypochromic anemia, and target cells may be seen |
|
|
Treatment of beta thalassemia major
|
Frequent packed RBC transfusions are necessary to sustain life. this can lead to iron overload and symptoms of CHF (similar to that seen in hemochromatosis). Therefore these patients are often treated with deferoxamine- a chelating agent that eliminates excess iron
|
|
|
Thalassemia minor
|
-aka heterozygous beta-chain thalassemia
- clinical features: patients are usually asymptomatic. - a mild microcytic, hypochromic anemia is the only symptom - Diagnosis: hemoglobin electrophoresis - treatment: usually not necessary - pts are not transfusion dependent |
|
|
4 types of alpha thalassemia
|
1. silent carriers- mutation/deletion of only ONE of the four alpha loci. Asx, normal Hgb and Hct, no tx
2. alpha thalassemia trait (or minor) - mutation/deletion of two alpha loci. Characterized by mild microcytic hypochromic anemia. Most common in african american patients. no treatment necessary. 3. Hb H disease - hgb Barts- mutation/deletion of THREE alpha loci. Results in hemolytic anemia and splenomegaly. There is significant microcytic, hypochromic anemia. Hgb Electrophoresis shows Hgb H. Treatment is ofteh nthe same for patients with b-thal major and splenectomy is sometimes helpful 4. Mutation/deletion of all FOUR alpha loci- This is either fatal at birth (hydrops fetalis) or shortly after birth |
|
|
Sideroblastic Anemia
1. What causes it? 2. Clinical findings 3. Treatment |
1. An abnormality in RBC metabolism. It can either be hereditary or acquired. Acquired causes include drugs (chloramphenicol, Isoniazid, alcohol), exposure to lead, collagen vascular disease, and neoplastic disease (myelodysplastic syndromes)
2. increased serum iron and ferritin, normal TIBC, TIBC saturation is normal/elevated, which distinguishes it from iron deficiency. You may see ringed sideroblasts in the bone marrow 3. Remove offending agents and consider pyridoxine (Vitamin B6) |
|
|
Anemia of chronic disease
1. It was clinical setting may you see it? 2. Lab findings 3. What type of anemia is it? 4. Treatment |
1. This occurs in the setting of chronic infection (TB, lung abscess, etc), cancer (e.g. lung, breast, Hodgkins), inflammation (RA, SLE, or trauma). The release of inflammatory cytokines has a suppressive effect on erythropoiesis. It may be difficult to differentiate between this and IDA
2. Labs- low serum iron, low TIBC, low serum transferrin levels. Serum FERRITIN IS INCREASED 3. The anemia is usually normocytic and normochromic, but may be microcytic and hypochromic as well 4. There is no specific treatment other than treating the underlying process. Do NOT give iron. The anemia is usually mild and well-tolerated. |
|
|
Aplastic Anemias - general characteristics
1. main problem 2. causes |
1. bone marrow failure leading to pancytopenia (anemia, neutropenia, thrombocytopenia)
2. Causes- idiopathic in most cases, radiation exposure, medications (chloramphenicol, sulfonamides, gold, carbamezapine), Viral infection (HPV, hep C, hep B, EBV, CMV, Herpes Zoster, HIV). Chemicals (benzene, insecticides) |
|
|
clinical features of aplastic anemia
|
- signs of anemia - fatigue and dyspnea
- signs and symptoms of thrombocytopenia (petechiae, easy bruising) - increased incidence of infections (due to neutropenia) - can transform into acute leukemia |
|
|
Diagnosis of aplastic anemia
|
1. Normocytic, normochromic anemia
2. Perform a bone marrow biopsy for definitive diagnosis- this reveals hypocellular marrow and the absence of progenitors of all three hematopoietic cell lines |
|
|
Treatment of aplastic anemia
|
1. Bone marrow transplantation
2. transfusion of PRBCs and platelets if necessary 3. treat any known underlying causes |
|
|
Pernicious Anemia
|
- special cases of Vitamin B12- macrocytic anemia cause by an autoimmune disorder resulting in inadequate production of intrinsic factor, which leads to impaired absorption of vitamin B12 in the terminal ileum
- perform Schilling test to determine if this is the cause - most common cause of vitamin b12 deficiency in the western world - may see hypersegmented neutrophils on blood smear |
|
|
Vitamin B12 deficiencies - general characteristics
1. what 2 important reactions is vit B12 important for 2. Vit b12 stores- where and how long can it last 3. where found in diet 4. absorption process |
1. Vitamin B12 is important in two important reactions (a. as a cofactor in the conversion of homocysteine to methionine, and b. as co-factor in the conversion of methylmalonyl CoA to succinyl CoA)
2. Vitamin B12 stores in the liver are plentiful and can sustain an individual for 3 or more years 3. The main dietary sources of Vitamin B12 are meat and fish 4. Vitamin B12 is bound to intrinsic factor (produced by gastric parietal cells) so it can be absorbed in the terminal ileum |
|
|
Causes of vitamin B12 deficiency
|
* almost all cases are due to impaired absorption
1. pernicious anemia (lack of intrinsic factor from gastric parietal cells) - most common cause in the western hemisphere 2. gastrectomy 3. poor diet ( e.g. strick vegetarianism), alcoholism 4. crohn's disease, ileal resection (terminal ileum - approx the last 100 cm) 5. other organisms competing for vit b12 (diphyllobrothrium latum infection (fish tapework) and blind-loop syndrome (bacterial overgrowth) |
|
|
Clinical features of vitamin b12 deficiency
|
1. anemia
2. sore tongue (stomatitis and glossitis) 3. Neuropathy - can distinguish between vitamin b12 and folate deficiency - demyelination of the posterior columns, in the lateral corticospinal tracts and spinocerebellar tracts-- leads to loss of position/vibratory sensation in the lower extremities, ataxia, and UMN signs (increased DTRs, spasticity, weakness, Babinski sign) - can lead to urinary and fecal incontinence, impotence - can lead to dementia - investigate in the workup for dementia |
|
|
Diagnosis of vitamin B12 deficiency
|
1. Peripheral Blood Smear- megaloblastic anemia (macrocytic RBCs with a MCV > 100). hypersegmented neutrophils
2. Serum B12 level is low (<100 pg/mL) 3. serum MMA and homocysteine levels - there are usually elevated in vitamin B12 deficiency and are useful if the vit b12 level is borderline 4. antibodies against instrinsic factor can help in the dx of pernicious anemia 5. perform a schilling test- provides information regarding the cause of the vit b12 deficiency a. Give an IM dose of unlabeled Vit B12 to saturate binding sites b. give an oral dose of radioactive vit B12: measure the amount of vit b12 in urine and plasma to determine how vit b12 was absorbed c. Repeat the test (oral radioactive B12) with the addition of intrinsic factor. If malabsorption is the problem, then adding the intrinsic factor will not do anything, but if pernicious anemia is present, adding IF will improve serum vit b12 levels |
|
|
Treatment of vit b12 deficiency and associated macrocytic anemia
|
1. parenteral therapy is preferred-- cyanocobalamine (vitamin B12) IM once per month
|
|
|
Folate deficiency - general characteristics
1. folic acid stores 2. source of folate |
1. Folic acid stores are limited. Inadequate intake of folate over a 3 month period is sufficient to cause a deficiency
2. green vegetables are the main source of folate. Overcooking of vegetables may remove the folate |
|
|
Causes of folate deficiency and corresponding macrocytic anemia
|
1. Inadequate dietary intake such as "tea and toast" (most common cause)
2. Alcoholism 3. long-term use of oral antibiotics 4. increased demand 5. pregnancy 6. hemolysis 7. use of folate antagonists such as methotrexate 8. anticonvulsant medications (phenytoin) 9. Hemodialysis |
|
|
Serum methylmalonic acid (MMA) and homocysteine levels in vitamin B12 and folate deficiencies respectively,
|
1. Vitamin B12 deficiency - increased MMA and Homocysteine levels
2. Folate deficiency- increased homocysteine levels but normal MMA levels |
|
|
Clinical features of folate deficiency and treatment
|
Anemia - fatigue etc
- NO neurologic deficits treatment- daily folic acid replacement |
|
|
Hemolytic Anemias- General Characteristics
1. definition 2. bone marrow response and pathophys 3. acute vs chronic |
1. Premature destruction of RBCs that may be due to a variety of causes
2. Bone marrow is normal and responds appropriately by increasing erythropoeisis leading to an increased retic count, but it cannot keep up and anemia results 3. Can be acute or chronic with a corresponding variation in clinical features |
|
|
Extrinsic causes of hemolytic anemias
|
- factor extrinsic to RBC defects lead to hemolysis
- most cases are acquired - immune hemolysis - mechanical hemolysis (prostethic heart valves, microangiopathic hemolytic anemia) - medications, burns, toxins (e.g. from a snake bite or brown recluse spider), infection (malaria, clostridium) and so on |
|
|
Intrinsic causes of hemolytic anemias
|
- hemolysis due to intrinsic RBC defects
- most cases are inherited - hemoglobin abnormality - sickle cell anemia, hemoglobin C disease, thalassemias - membrane defects - hereditary spherocytosis, paroxsymal nocturnal hemoglobinuria (PNH) - enzyme defects- G6PD deficiency, pyruvate kinase deficiency |
|
|
Classification of hemolytic anemias based on the predominant site of hemolysis
|
1. intravascular hemolysis- within the circulation
2. extravascular hemolysis - within the reticuloendothelial system- primarily in the spleen |
|
|
Clinical features of hemolytic anemias
|
1. Signs and symptoms of anemia
2. signs and symptoms of underlying disease (e.g. bone crises in sickle cell disease) 3. Jaundice 4. dark urine color (due to hemoglobinuria, not bilirubin) may be present. This indicates an intravascular process 5. Hepatosplenomegaly (HSM), cholelithiasis, lymphadenopathy (in chronic cases) |
|
|
Diagnosis of hemolytic anemias
|
1. hgb/hct level depends on degree of hemolysis and reticulocytosis
2. elevated retic count due to increased RBC production 3. peripheral smear - schistocytes suggest intravascular hemolysis (trauma or mechanical hemolysis) - spherocytes or helmet cells suggest extravascular hemolysis (depending on the cause) - sickled RBCs- sickle cell anemia - Heinz bodies in G6PD deficiency 4. Haptoglobin levels are low in hemolytic anemias (especially in intravascular hemolysis). Haptoglobin binds to hemoglobin, so its absence means the hemoglobin was destroyed 5. LDH level is elevated - LDH is released when RBCs are destroyed 6. Elevated indirect (unconjugated) bilirubin due to degradation of heme because RBCs are destroyed 7. Direct coombs test (detects antibody or complement on RBC membrane) 8. osmotic fragility test |
|
|
treatment of hemolytic anemia
|
1. treat the underlying cause
2. transfusion of PRBCs if severe anemia is present or patient is hemodynamically compromised 3. folate supplements (folate is depleted in hemolysis) |
|
|
Causes of sickle cell anemia
|
1. Autosomal recessive disorder the results when the normal Hemoglobin A is replaced with a mutant Hgb S. Sickle cell disease is caused by an inheritance of Hgb S genes (homozygous)
2. Hgb S may be distinguished from Hgb A by electrophoresis because of the substitution of an uncharged valine for a negatively charged glutamic acid at the 6th position of the beta-chain 3. under reduced oxygen conditions (acidosis, hypoxia, changes in temperature, dehydration and infection) the hgb molecules polymerize, causing the RBCs to sickle. Sickled RBCs obstruct small vessels leading to ischemia. Almost every organ can be effected (heart, brain, lungs, kidneys, eyes, genitals etc) |
|
|
Sickle cell trait
|
1. about 1 in 12 people of African decent carry the sickle cell trait, they are heterozygous. The sickle cell trait also appears in Italians, Greeks, and Saudi Arabians
2. Patients with sickle cell trait are not anemia and have a normal life expectancy 3. screening can identify asymptomatic carriers (sickle cell trait) for whom genetic counseling can be provided |
|
|
Prognosis of sickle cell anemia
|
1. survival correlates with the frequency of vaso-occlusive crises- more frequent crises are associated with a shorter lifespan
2. if there are more than 3 crises per year then the median age of death is 35 years. Patients with fewer crises per year may live into their 50s 3. in general, sickle cell disease reduces life expectancy by 25-30 years |
|
|
Clinical features of sickle cell disease that are due the severe, lifelong hemolytic anemia
|
1. jaundice, pallor
2. gallstone disease (very common)- pigmented gallstones 3. the anemia itself is well compensated and is rarely transfusion dependent 4. high output heart failure may occur over time (secondary to anemia) - many adults eventually die of CHF 5. aplastic crises- there are usually provoked by a viral infection such as Human parvovirus B19, which reduces the ability of bone marrow to compensate. tx is with blood transfusion. Patient usually recovers within 7-10 days |
|
|
clinical features of sickle cell anemia that are secondary to vaso-occlusion
|
1. Painful crises involving the bone- bone infarction causes severe pain. This is the most common clinical manifestation. Bone main usually involves multiple sites (tibia, humerus, femur). It may be bilateral. The pain is self-limiting and usually lasts 2-7 days
2. Hand-foot syndrome (dactylitis) - painful swelling of the dorsa of hands and feet seen in infancy and early childhood (usually 4-6 months). Often the first manifestation of the disease). Caused by avascular necrosis of the metocarpal and metatarsal bones 3. Acute chest syndrome - due to repeated episodes of pulmonary infarctions. clinical presentation is similar to pneumonia. Associated with chest pain, respiratory distress, pulmonary infiltrates, and hypoxia 4. repeated episodes of splenic infarctions- these lead to autosplenectomy as the spleen is reduces to a small, calcified remnant. The spleen is large in childhood but no longer palpable by age 4 5. avascular necrosis of joints - most common in hip and shoulder |
|
|
clinical features of sickle cell anemia that are secondary to vaso-occlusion continued..
|
6. Priapism- erection due to vaso-occlusion, usually lasting between 30 min and 3 hours. Usually subsides spontaneously after urine is passed, light exercise or a cold shower. A trial of hydralazine or nifedipine or use of an antiandroen (stilbestrol) may prevent future episodes. Sustain priapism (lasting more than 3 hours is rare- less than 2% but is a medical emergency)
7. CVAs- the result of cerebral thrombosis, primarily affects children 8. Ophthalmologic complications - retinal infarcts, vitreous hemorrhage, proliferative retinopathy, retinal detachment 9. Renal papillary necrosis with hematuria- usually painless - a common complication in up to 20% of patients. Seldom requires hospitalization and may cease spontaneously. 10. chronic leg ulcers due to vaso-occlusion (decreased blood flow to superficial vessels- typically over the lateral malleoli 11. abdominal crisis may occur in adulthood- mimics acute abdomen |
|
|
infectious complications of sickle cell disease
|
1. functional asplenia results in increased susceptibility to infections (particularly encapsulated bacteria such as H. flu, S, pneumo)
2. predispostion to salmonella osteomyelitis - also due to splenic malfunction - there is also delayed growth and sexual maturation, especially in boys |
|
|
Diagnosis of Sickle Cell Anemia
|
1. Anemia is the most common finding
2. Peripheral smear- sickle-shaped RBCs 3. Hemoglobin electrophoresis is required for diagnosis. In most cases, diagnosis is made from newborn screening tests |
|
|
Treatment of sickle cell anemia
|
1. Advise the patient: Avoid high altitudes (low O2 tension can precipitate crises), maintain fluid intake, treat infections promptly
2. early vaccination for H. flu, S. pneumo, and N. meningitidis 3. Prophylactic penicillin for children until 6 years of age- start at 4 months 4. folic acid supplements - due to chronic hemolysis 5. Management of painful crises- hydration (oral if mild, otherwise IV NS), morphine for pain control, keep patient warm, supp O2) 6. Hydroxyurea - enhances hgB F levels, which interferes with the sickling process, results in reduced incidence of painful crises, accelerates healing of leg ulcers and may reduce recurrence 7. blood transfusion - not used unless absolutely necessary. Base the need for transfusion on the patient's clinical condition and not the Hgb levels. Transfusion should be considered in acute chest syndrome, stroke, priapism that does not respond to fluids/analgesia, and cardiac decompensation 8. BMT and gene therapy? |
|
|
Hereditary spherocytosis - general characteristics
|
1. AD inheritance of a gene coding for spectrin and other RBC proteins. Spectrin content is decreased but not totally absent.
2. There is a loss of RBC membrane surface area without reduction in RBC volume, necessitating a spherical shape. The spherical RBCs become trapped and destroyed by the spleen (macrophages)- extravascular hemolysis |
|
|
Clinical features of hereditary spherocytosis
|
1. hemolytic anemia (can be severe)
2. jaundice 3. splenomegaly 4. gallstones 5. occasional hemolytic crises |
|
|
Diagnosis of hereditary spherocytosis
|
1. RBC osmotic fragility to hypotonic saline
a. tests the ability of RBCs to swell in graded series of hypotonic solutions. b. because of their shape, spherocytes tolerate less swelling before they rupture; thus they are osomotically fragile. The RBCs undergo lysis at higher (thus earlier) oncotic pressure 2. Elevated retic count, elevated MCHC 3. Peripheral blood smeat would reveal spherocytes 4. Direct coombs test would be negative. This is helpful in distinguishing this disease from autoimmune hemolytic anemia, in which spherocytes may also be seen *spherocyte- bi-concave disk with no central palor |
|
|
Treatment of hereditary spherocytosis
|
Splenectomy
|
|
|
Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD def) - general characteristics
|
1. X-linked recessive disorder that primarily affects men
2. known precipitants include sulfonamides, nitrofurantoin, primaquin, dimercaperol, fava beans and infection |
|
|
Types of G6PD deficiency (2)
|
1. A mild form is present in 10% of African American men (A-variant)- in this form, hemolytic episodes are usually self-limited because they mainly involve only the older RBCs and spare the younger RBCs (the younger RBCs have sufficient G6PD to prevent RBC destruction). Hemolytic episodes are usually triggered by infection or by drugs such as antimalarials (primaquine) and sulfur-containing antibiotics (Bactrim etc)
2. A more sever form is present in people of Mediterranean descent. In this form, young as well as old RBCs are G6PD deficient. Causes severe hemolytic anemia when exposed to fava beans. May require transfusions until the drug is eliminated from the body |
|
|
Clinical features of G6PD deficiency
|
1. Episodic hemolytic anemia that is usually drug-induced
2. dark urine and jaundice on physical exam |
|
|
Diagnosis of G6PD deficiency
|
1. Peripheral blood smear
- shows "bite cells" - RBCs after the removal of the heinz bodies look as if they have had bites taken out of them. The "bitten" areas are secondary to phagocytosis of heinz bodies by splenic macrophages - heinz bodies "howell jolly bodies" (abnormal hemoglobin precipitates within RBCs) are visible with special stains 2. deficient NADPH formation on G^PD assay 3. measurement of G6PD levels is diagnostic, however these may be normal during a hemolytic episode because the RBCs that are most deficient have already been destroyed. Repeat the testing after the episode to confirm |
|
|
Treatment of G6PD deficiency
|
1. Avoid drugs that precipitate hemolysis
2. maintain hydration 3. Perform RBC transfusion when necessary |
|
|
Autoimmune Hemolytic Anemia (AIHA)- general characteristics
1, pathophys 2. what determines prognosis, site of RBC destruction and response to treatment? 3. clinical course |
1. Production of autoantibodies toward RBC membrane antigen(s) which leads to destruction of these RBCs
2. The type of antibody produced (immunoglobulin IgG or IgM) determines the prognosis, site of RBC destruction and response to treatment 3. The course is variable but tends to be more fulminant in children than in adults. |
|
|
Warm Autoimmune Hemolytic Anemia (AIHA)
1. what is the antibody 2. site of hemolysis 3. causes of hemolysis |
1. Autoantibody is IgG, which binds optimally to the RBC at 37 deg C -- hence "warm"
2. results in extravascular hemolysis- the primary site of RBC sequestration is the spleen. Splenomegaly is a common feature 3. causes - primary (idiopathic), secondary to lymphomas, leukemias (CLL) other malignancies, collagen vascular diseases (especially SLE) and drugs such alpha-methyldopa- aldomet- a2 agonist) |
|
|
Cold Autoimmune Hemolytic Anemia
|
1. Autoantibody is IgM, which binds optimally to the RBC membrane at cold temperatures (usually 0 to 5 deg C)
2. Produces complement activation and intravascular hemolysis - primary site of RBC sequestration is the liver 3. Causes- can be idiopathic (elderly) or due to infection (such as mycoplasma pneumonia infection or infectious mononucleosis) |
|
|
Clinical features of Autoimmune hemolytic anemia
|
1. signs and symptoms of anemia (fatigue, pallor)
2. jaudice if significant hemolysis is present 3. features of the underlying disease |
|
|
Diagnosis of autoimmune hemolytic anemia (AIHA)
|
1. Direct Coombs test
- if RBCs are coated with IgG (positive direct Coombs test) then the diagnosis is warm AIHA - if the RBCs are coated with complement alone, then the diagnosis is cold AIHA 2. If there is a positive cold agglutinin titer, then the diagnosis is cold AIHA 3. spherocytes may be present in warm AIHA |
|
|
Treatment of autoimmune hemolytic anemia
1. mild 2. warm 3. cold |
1. often, no treatment is necessary in either type of AIHA, because the hemolysis is mild. If it is more severe, the therapeutic approach depends on the type of autoantibody causing the hemolysis
2. Warm AIHA - glucocorticoids are the mainstay of therapy- splenectomy is used for patients whose condition does not respond to glucocorticoids. Immunosuppresion (azathioprine or cyclophosphamide) may be beneficial. RBC transfusions if absolutely necessary. Folic acid supplements 3. Cold AIHA - avoiding exposure to the cold prevents bouts of hemolysis and anemia. RBC transfusions if absolutely necessary. Various chemotherapy agents. Steroids are NOT beneficial |
|
|
Paroxsymal Nocturnal Hemoglobinuria (PNH)- general characteristics
1. definition 2. cause |
1. an acquired disorder that affects hematopoietic stem cells and cells of all blood lineages
2. This is causes by a deficiency of anchor proteins that link-complement-inactivating proteins to the blood cell membranes. The deficiency of this anchoring mechanism results in an unusual susceptibility to complement-mediated lysis of RBCs, WBCs and platelets |
|
|
Clinical features of paroxsymal nocturnal hemoglobinuria
|
1. Chronic intravascular hemolysis- results in paroxysmal hemoglobinuria, elevated LDH
2. normochromic normocytic anemia (unless iron deficiency is present) 3. Pancytopenia 4. Thrombosis of venous systems can occur (e.g. of the hepatic veins-- budd-chiari syndrome) 5. may evolve into aplastic anemia, myelodysplasia, myelofibrosis, and acute leukemia 6. abdominal, back and MSK pain |
|
|
Diagnosis of paroxsymal nocturnal hemoglobinuria (PNH)
|
1. Ham's test - the patient's cell are incubated in acidified serum, triggering the alternative complement pathway, resulting in lysis of PNH cells, but not normal cells.
2. Sugar water test- the patient's serum is mixed in sucrose. In PNH, hemolysis ensues 3. flow cytometry of anchored cell surface proteins (CD55 and CD 59) - much more sensitive and specific for PNH |
|
|
Treatment of paroxsymal nocturnal hemoglobinuria (PNH)
|
1. Glucocorticoids (prednisone) are the usual initial therapy, but many patients do not respond
2. bone marrow transplantation |
|
|
Thrombocytopenia - general characteristics
1. definition 2. Causes |
1. platelet counts <150,000. Normal is 150,000 - 400,000
2. Causes - a. decreased production- bone marrow failure, acquired (aplastic anemia), congenital (Fanconi's anemia), congenital intrauterine rubella. Bone marrow invasion (tumors, leukemia, fibrosis). Bone marrow injury: drugs (ethanol, gold, cancer chemo agents- chloramphenicolm chemicals (benzene), radiation, infection - increased destruction: Immune (infection, drug-induced, immune thrombocytopenic purpura ITP, SLE, heparin induced thrombocytopenia (HIT) type 2, HIV associated thrombocytopenia. Non-immune: disseminated intravascular coagulation (DIC), thrombotic thrombocytopenic purpura (TTP), HIT type 1 - sequestration from splenomegaly - dilutional - after transfusions or hemorrhage - pregnancy- usually an incidental finding (esp in 3rd trimester) but can also occur in the setting of pre-eclampsia or eclampsia (HELLP syndrome - hemolysis, elevated liver enzymes and low platelets of pregnancy) |
|
|
Diagnosis of thrombocytopenia
|
1. CBC - platelet count
2. bleeding time, prothrombin time (PT) and partial thromboplastin time (PTT) 3. to determine the cause of thrombocytopenia, the following may be helpful: examination of peripheral blood smear, bone marrow biopsy |
|
|
Clinical features of thrombocytopenia
|
1. cutaneous bleeding: petechiae (most common in dependent areas), confluent petechiae are called purpura, ecchymoses at sites of minor trauma
2. mucosal bleeding: epistaxis, menorrhagia, hemoptysis, bleeding in GI and GU tracts 3. Excessive bleeding after procedures or surgery 4. Intracranial hemorrhage and heavy GI bleeding can be life-threatening and can occur when platelets are severely low 5. unlike coagulation disorders (e.g. hemophilia), heavy bleeding into tissues and joints (hemarthroses, hematomas) is NOT seen in thrombocytopenia |
|
|
Treatment of thrombocytopenia
|
1. treat the underlying cause
2. platelet infusion- use depending on the cause and severity of thrombocytopenia 3. discontinue NSAIDs and other antiplatelet agents, and anticoagulants |
|
|
Severity of thrombocytopenia and associated risk
1. > 100,000 2. 20,000 - 70,000 3. < 20,000 4. < 5,000 |
1. > 100,000 - abnormal bleeding (even after trauma or surgery) is unusual
2. 20,000- 70,000 - increased bleeding hemorrhage during surgery or trauma 3. <20,000 - minor spontaneous bleeding: easy bruising, petechiae, epistaxis, menorrhagia, bleeding gums 4. < 5,000 - major spontaneous bleeding: intracranial bleeding, heavy GI bleeding |
|
|
Immune (Idiopathic) Thrombocytopenic Purpura - general characteristics
1. pathophys 2. two forms - who do they usually occur in and what is the course of the disease generally like? |
1. This results from autoimmune antibody formation against host platelets. These antiplatelet antibodies (IgG) coat and damage platelets, which are then removed by splenic macrophages (RES binds self-immunoglobulins attached to the platelet)
2. Occurs in two forms a. acute form - seen in children, preceded by a viral infection (in most cases), usually self-limited - 80% resolve spontaneously within 6 months b. chronic form- usually seen in adults, most commonly in women between 20-40 years of age. Spontaneous remissions are rare |
|
|
Clinical features of ITP - immune/idiopathic thrombocytopenic purpura
|
1. Petechiae and ecchymoses on the skin- many patients will have only very minimal bleeding symptoms despite extremely low platelet counts (< 5,000)
2. bleeding of the mucous membranes 3. NO splenomegaly |
|
|
Diagnosis of Immune/Idiopathic Thrombocytopenic Purpura (ITP)
|
1. The platelet count is frequently less than 20,000. The remainder of the blood count is normal
2. peripheral smear shows decreased platelets 3. bone marrow aspiration shows increased megakaryocytes (platelet precursor cells) 4. increased amount of platelet-associated IgG |
|
|
Treatment of Idiopathic/Immune Thrombocytopenic Purpura
|
1. Adrenal corticosteroids
2. IV immune globulin- saturates the reticulo-endothelial system binding sites for platelet-bound self-immunoglobulin so there is less platelet uptake and destruction by the spleen 3. splenectomy- induces remission in 70-80% of the cases of chronic ITP 4. platelet transfusions- for life-threatening and serious hemorrhagic episodes 5. Two new drugs - romiplastin and eltrombopag, have been approved for splenectomy-resistant patients. Both of these drugs work as thrombopoeitin receptor agonists to increase platelet production |
|
|
Thrombotic Thrombocytopenic Purpura (TTP) - general characteristics
|
1. TTP is a rare disorder of platelet consumption. The cause is unknown
2. hyaline microthrombi (mostly platelet thrombi) occlude small vessels- any organ may be involved. They cause mechanical damage to RBCs (schistocytes on peripheral smear) 3. this is a LIFE THREATENING EMERGENY that is responsive to therapy . If untreated, death occurs within a few months |
|
|
Clinical features of thrombotic thrombocytopenic purpura
|
1. hemolytic anemia (microangiopathic)
2. thrombocytopenia 3. Acute renal failure (mild) 4. fever 5. fluctuating, transient neurologic signs- can range from mental status change to hemiplegia |
|
|
Treatment of thrombotic thrombocytopenic purpura (TTP)
|
1. Plasmapheresis (large volume) - begin as soon as the diagnosis is established (delay in treatment is life-threatening)
- response is usually good (monitor platelet count, which should increase) 2. corticosteroids and splenectomy- may be of benefit in some cases 3. platelet transfusions are contraindicated |
|
|
Thrombotic thrombocytopenic purpura (TTP) vs hemolytic uremic syndrome (HUS)
|
TTP - there is no consumption of the clotting factors in TTP, so PT and PTT are normal
- TTP = HUS + fever + AMS - HUS = microangiopathic hemolytic anemia + thrombocytopenia + renal failure |
|
|
Heparin-Induced Thrombocytopenia (HIT)
1-3. risk, definition, diagnosis 4. specific diagnostic test 5. treatment |
1. can occur with any amount of heparin. Mostly occurs with unfractionated heparin. LMWH has a much lower risk of HIT.
2. drop in platelets a few days after administration of heparin. Platelets aggregate "clump" leading to venous thrombosis (DVT and subsequent PE) 3. decrease in platelet count by 50% after giving heparin suggests HIT 4. Diagnostic tests: antiplatelet factor IV antibody or serotonin release assay 5. Treatment: stop heparin. If anticoagulation is indicated (venous thrombosis) then give a direct thrombin inhibitor such as lepirudin ( hirudin comes from leeches) |
|
|
Bernard-Soulier Syndrome
|
- autosomal recessive disease
- disorder of platelet adhesion (to subendothelium) due to deficiency of platelet glycoprotein Ib-IX - on peripheral smear, PLATELETS APPEAR ABNORMALLY LARGE - platelet count is mildly low - defect in platelet to collagen adhesion (GPIb binds vWF) |
|
|
Glanzmann's Thrombasthenia
|
- autosomal recessive disease
- disorder of platelet to platelet aggregration due to deficiency of GPIIb-IIIa - defect in platelet plug formation - bleeding time is prolonged - platelet count is normal - blood smear shows NO platelet clumping |
|
|
What is the main complication of Heparin Induced Thrombocytopenia (HIT)
|
DVT and PE
|
|
|
Diagnosis of TTP
|
- decreased platelet survival leads to increased bleeding time and decrease platelet count
- deficiency in ADAMTS 13 (vWF metalloprotease) --> decreased degradation of vWF multimers - pathogenesis - Increased large vWF multimers--> increased platelet aggregation and thrombosis - schitocytes and inc LDH |
|
|
What type of bleeding problem might you see in uremia?
|
A qualitative platelet defect with normal platelet count, PT and PTT, but an increased bleeding time.
- dialysis can improve this |
|
|
Howell-Jolly Bodies
|
- basophilic nuclear remnants found in RBCs.
- these are seen in patients with functional hyposplenia or asplenia |
|
|
von Willebrand's Disease (vWD) - general characteristics
1. definition 2. role of vWF 3. how common 4. three subtypes |
1. Autosomal dominant disorder characterized by deficiency or defect of factor VIII-related antigen (vWF)
2. vWF enhances platelet aggregation and adhesion (the first steps in clot formation) 3. This is the most common inherited bleeding disorder (affects 1-3% of the population) 4. There are three major subtypes - Type 1- the most common form decreases levels of vWF, Type 2 is less common and exhibits qualitative abnormalities of vWF, type 3 is the least common and involves absent vWF (very severe disease) |
|
|
Clinical features of vWD
|
1. Cutaneous and mucosal bleeding- epistaxis, easy bruising, excessive bleeding from scratches and guts, gingival bleeding
2. Menorrhagia (affects more than 50% of woman with vWD) 3. GI bleeding is possible |
|
|
Diagnosis of von Willebrand Disease
|
1. Diagnosis is derived from clinical findings and laboratory information, which can be variable
2. prolonged bleeding time (but normal platelet count) - PTT may be prolonged (because of associated factor VIII deficiency) but a normal PTT does not exclude this 3. decreased plasma vWF, decreased factor VIII activity 4. reduced ristocetin-induced platelet aggregation (ristocetin assay) |
|
|
Treatment of vWD
|
1. DDAVP (desmopressin)-- induces endothelial cells to secrete vWF
a. treatment of choice for type 1 vWD (the most common type) b. some patients with type 2 vWD may respond to DDAVP, but it is not effective in type 3 2. Factor VIII concentrates (containing high-molecular weight vWF)- give to all patients with vWD (any type) after major trauma or during surgery. Recommended for type 3 vWD and type 2 patients who do not respond to desmopressin 3. cryoprecipitate is NOT recommended as a treatment for vWD because it carries the risk of viral transmission 4. avoid aspirin/NSAIDs as well as IM injections (exacerbates bleeding tendency) |
|
|
How does the bleeding in vWD compare to the bleeding in hemophilia?
|
in general, the bleeding in vWD is much milder than in hemophilia. Spontaneous hemarthroses do NOT occur (because it is primarily a platelet problem)
- in many patients the disease is so mild that is not diagnosed until the time of surgery or trauma |
|
|
Hemophilia A- general characteristics
1. inheritance pattern |
1. X-linked recessive disorder - affects male patients primarily
2. caused by deficiency or defect of factor VIII coag protein 3. Bleeding tendency is related to factor VIII activity |
|
|
Clinical features of Hemophilia A
|
1. Hemarthrosis - knees are the most common site, but any joint can be involved. Progressive joint destruction can occur secondary to recurrent hemarthroses. Maintaining normal factor VIII levles (by prophylactic admin of factor VIII concentrate) can minimize joint destruction. Syovectomy (arthroscopic) may be needed
2. Intracranial bleeding - second most common cause of death - AIDS from transfusion prior to screening is the most common cause of death. any head trauma is potentially life threatening and requires urgent eval. 3. Intramuscular hematomas 4. retroperitoneal hematomas 5. hematuria or hemospermia |
|
|
Diagnosis of Hemophilia A
|
1. prolonged PTT
2. low factor VIII level with a normal level of vWF |
|
|
Treatment of acute hemarthrosis in hemophilia A
|
1. analgesia (codeine with or without acetaminophen)- avoid aspirin and NSAIDs
2. immobilization of the joint, ice packs, non-weight bearing |
|
|
Treatment of Hemophilia A
|
1. Clotting factor replacement
a. factor VIII concentrate is the mainstay of therapy (both plasma-derived and recombinant factor VIII are available) - for acute bleeding episodes and before surgery or dental work b. cryoprecipitate and fresh frozen plasma (FFP) are NOT recommended because of the risk of viral transmission 2. DDAVP (desmopressin) - may be helpful in patients with mild disease. It can increase the levels of factor VIII up to 4-fold 3. Gene therapy is a possible future treatment |
|
|
vWF and vWD
1. site of synthesis 2. functions 3. inheritance pattern 4. levels in vWD and hemophilia |
1. endothelial cells and megakaryocytes (platelet precursor cells)
2. functions- platelet adhesion- mediates the adhesion of platelets to injured vessel walls (i.e. it reacts with platelet GPIb/IX and subendothelium). Binds the factor VIII and protects it from degradation 3. autosomal dominant 4. low in vWD and normal in hemophilia |
|
|
Factor VIII
1. site of synthesis 2. function 3. inheritance of hemophilia 4. levels in vWD and hemophilia |
1. liver
2. fibrin clot formation 3. x-linked recessive 4. vWD- reduced, hemophilia- very low |
|
|
Hemophilia B - general characteristics
|
1. caused by a deficiency of factor IX
2. x-linked recessive disorder 3. much less common than hemophilia A 4. clinical features are identical to those of hemophilia A 5. treatment involves administration of factor IX concentrates. Desmopressin does NOT play a role in treatment. |
|
|
Disseminated Intravascular Coagulation (DIC) - general characteristics
1. what is it? 2. what patient population is it most common in? 3. course |
1. DIC is characterized by abnormal activation of the coagulation sequence, leading to formation of microthrombi throughout the microcirculation. This causes consumption of platelets, fibrin, and coagulation factors. Fibrinolytic mechanisms are activated, leading to hemorrhage. Therefore, bleeding and thrombosis occur simultaneously
2. Most common in critically ill patients (in ICU) but can occur in healthy patients as well 3. Can be acute and fatal or more gradual |
|
|
Causes of DIC
|
1. Infection - most common cause, especially in cases of gram-negative sepsis, but any infection can cause DIC
2. obstetric complications (placenta and uterus have increased tissue factor)-- amniotic fluid emboli (often acute and fatal), retained dead fetus (often chronic), abruptio placentae 3. Major tissue injury- trauma, major surgery, burns, fractures 4. malignancy- lungs, pancreas, prostate, GI tract, acute promyelocytic leukemia 5. shock, circulatory collapse 6. snake venom (rattlesnakes) |
|
|
Clinical features of DIC
|
1. bleeding tendency (more common in acute cases)
a. superficial hemorrhage (ecchymoses, petechia and purpura) b. bleeding from the GI tract, GU tract, gingival or oral mucosa c. oozing from sites of procedures, incisions and so on 2. Thrombosis- occurs most often in chronic cases. End-organ infarction may develop. All tissues are at risk, especially the CNS and kidneys |
|
|
Diagnosis of DIC
|
1. Increased PT, PTT, bleeding time
2. increase fibrin split products (due to activation of the fibrinolytic system) 3. D-dimer 4. Decreased fibrinogen level - a normal or elevated level essentially rules out DIC 5. decreased platelet count (thrombocytopenia) 6. peripheral blood smear reveals schistocytes from damage of the RBCs as they go through the microcirculation (with microthrombi) |
|
|
Treatment of DIC
|
1. Management of the condition the precipitated DIC
2. Supportive measures may be indicated if severe hemorrhage is present (these are only temporizing measures) a. FFP replaces all the clotting factors b. platelet transfusions c. cryoprecipitate replaces clotting factors and fibrinogen d. low doses of heparin (IV or SC) inhibit clotting and can prevent consumption of the clotting factors. The use of heparin is controversial, given in only rare cases in which thrombosis dominates the clinical picture e. other supportive measures include oxygen and IV fluids. Maintain BP and renal perfusion |
|
|
Vitamin K deficiency - general characteristics
1. factors that depend on vitamin K 2. sources of vitamin K |
1. Several clotting factors depend on vitamin K as a co-factor in their synthesis by the liver (factors II, VII, IX, and X, protein C and S). the process is post-translational modification (gamma-carboxylation)
2. Sources of vitamin K include diet (e.g. leafy green vegetables) and synthesis by intestinal bacterial flora |
|
|
Causes of vitamin K deficiency
|
1. broad-spectrum antibiotics (suppression of gut flora) in patients who are NPO (inadequate dietary intake)
2. patients on TPN (unless vitamin K is added) 3. malabsorption of fat soluble vitamins (small bowel disease, IBD, obstructive jaundice, pancreatic insufficiency) 4. warfarin use- vitamin K antagonists- inhibits the gamma-carboxylation of vitamin K dependent clotting factors. - inactive clotting factors are made |
|
|
What vitamins are fat-soluble?
|
1. Vitamins D, A , K and E
|
|
|
Clinical features of Vitamin K deficiency
|
1. hemorrhage - serious bleeding can develop
2. PT is initially prolonged (factor VII has the shortest half-life). PTT prolongation follows (as other factors diminish) |
|
|
Treatment of Vitamin K deficiency
|
1. Vitamin K replacement (oral or SubQ)- it may take a few days for PT to return to normal
2. If bleeding is severe and emergency treatment is necessary, FFP should be transfused |
|
|
Coagulopathy of the Liver - general characteristics
|
1. All clotting factors are produced by the liver (except vWF)
2. Liver disease must be severe for coagulopathy to develop. Therefore, if the coagulopathy is due to liver failure, the overall prognosis for the patient is poor |
|
|
Why does coagulopathy develop in liver failure? (3)
|
1. There is decreased synthesis of clotting factors
2. cholestasis leads to decreased vitamin K absorption, which leads to vitamin K deficiency 3. hypersplenism (splenomegaly due to portal hypertension) causes thrombocytopenia |
|
|
Clinical features of coagulopathy due to liver failure
|
1. abnormal bleeding- GI bleeding is the most common, primarily due to varices secondary to portal hypertension, but exacerbated by coagulopathy
2. prolonged PT and PTT (especially PT) |
|
|
Treatment of coagulopathy due to liver failure
|
1. FFP (contains all clotting factors) if PT and PPT are prolonged or if bleeding is present
2. Vitamin K in certain cases (cholestasis) 3. platelet transfusion if thrombocytopenia is present 4. cryoprecipitate- if there is deficiency of fibrinogen |
|
|
Antithrombin (AT) III deficiency
|
1. autosomal dominant inheritance
2. AT III is an inhibitor of thrombin, so a deficiency leads to increased thrombosis |
|
|
Antiphospholipid antibody syndrome
|
1. Acquired hypercoaguability state
2. can present with arterial or venous thrombosis, recurrent fetal loss, or thrombocytopenia 3. the antibody may be against lupus anticoagulants, anticardiolipin, and B2 microglobulin among others |
|
|
Protein C deficiency
|
1. autosomal dominant inheritance
2. protein C is an inhibitor of factors V and VIII, so a deficiency leads to unregulated fibrin synthesis |
|
|
Protein S deficiency
|
Protein S is a co-factor of protein C, so a deficiency leads to decreased protein C activity
|
|
|
Factor V Leiden
|
- activate protein C resistnace
- a mutation in factor V gene - protein C can no longer inactivate factor V- leading to unregulated prothrombin activity and thus an increase in thrombotic events - the most common inherited cause of hypercoaguability |
|
|
Clinical features of inherited hypercoaguable states
|
1. Venous thromboembolisms (DVT and PE) are the most common sequelae. Such hypercoaguable disorders are usually not diagnosed until the patient has had several episodes of DVT or PE
2. Suspect an inherited hypercoaguable state if one or more of the following are present : The patient has a family history of DVT, Pe or thrombotic events, the patient has a history of this themselves, the patient's first thrombotic event was before age 40, the patient experience thrombosis in unusual sites (mesenteric veins, IVC, renal veins, or cerebral veins) |
|
|
Diagnosis of inherited hypercoaguability
|
- functional assays are available for antithrombin, antiphospholipid antibodies, protein C, protein S, factor V leiden, prothrombin gene mutation, and hyperhomocysteinemia
|
|
|
Treatment of inherited hypercoaguabilities
|
1. standard treatment of DVT or PE in patients without primary hypercoaguable states
2. Patients with any of these disorders who have had two or more thromboembolic events should be permanently anticoagulated with warfarin |
|
|
Heparin - mechanism of action
|
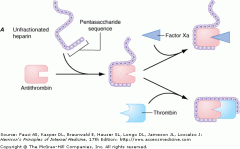
1. Potentiates the action of antithrombin to primarily inhibit clotting factors IIx and Xa
2. Prolongs PTT 3. Half life of standard heparin is 1 hour. It is longer for LMWHs- longer than 3 hours and up to 24 hours depending on which product |
|
|
Indications for heparin use
|
1. Venous thromboembolism: DVT or PE
2. Acute coronary syndromes: unstable angina or myocardial infarction 3. Low-dose standard heparin or LMWH for DVT prophylaxis 4. Atrial fibrillation in acute setting 5. After vascular bypass grafting |
|
|
Administration of heparin
1. standard heparin 2. LMWH |
1. A therapeutic dose is usually given IV, initiated with a bolus of 70-80 U/kg and followed by a continuous infusion (15 - 18 U/kg/hr). Therapeutic PTT is usually 60-90 seconds, but this varies depending on the clinical situation
- therapeutic heparin is now often monitored using antifactor Xa levels - a prophylactic dose is given subQ- low dose heparin (5,000 U SQ every 12 hours) PTT monitoring is not needed with SQ dosing 2. LMWH- therapeutic dose- given as a weight-adjusted dose. Prophylactic dose varies depending on the product. No need to monitor PTT |
|
|
Adverse effects of heparin
|
1. bleeding
2. Heparin induced thrombocytopenia (HIT) 3. possible osteoporosis- lower incidence with LMWHs 4. transient alopecia 5. rebound hypercoaguability after removal due to depression of ATIII |
|
|
Contraindications to Heparin use
|
1. Previous HIT
2. active bleeding, GI or intracranial 3. hemophilia, thrombocytopenia 4. severe HTN 5. recent surgery on eyes, spine or brain - recent trauma? |
|
|
Reversal of the effect of heparin or LMWH
|
1. The half-life of standard heparin is short, so it will cease to have an effect within 4 hours of its cessation
2. one can give protamine sulfate to reverse the effects of heparin if necessary (effectiveness is not proven, but it is the only potential antidote that exists in the case of severe bleeding) 3. LMWH has a longer half-life than standard heparin so it takes longer for the effects to fade |
|
|
Low-Molecular Weight Heparin - mechanism of action
|
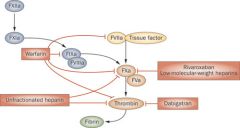
1. LMWHs mostly inhibit factor Xa (same as standard heparin), but there is less inhibition of factor IIa (thrombin) and platelet aggregation
2. they cannot be monitored by PT or PTT because they do not affect either 3. examples: enoxaparin, dalteparin, tinzaparin |
|
|
Low-Molecular Weight Heparin - Indications for use
|
1. LMWHs are being used more now because of their greater convenience compared with standard heparin, as well as decreased risk of SE (HIT and osteoporosis). They are given SQ, there is no PTT monitoring, and they are easier to give outpatient (can continue on this until warfarin is therapeutic)
2. excretion via kidneys- use cautiously if there is renal dysfunction 3. It is much more expensive than standard heparin, but often more cost-effective in the long run due to reduced testing, nursing time and length of hospital stay |
|
|
Warfarin - mechanism of action
|
1. a Vitamin K antagonist- leads to decrease in vitamin K dependent clotting factors (II, VII, IX and X) and protein C and S
2. Causes prolongation of PT and an increase in INR 3. It takes 4-5 days for the anticoagulant effect to begin. Therefore, start heparin as well if the foal is acute anticoagulation because heparin has an immediate effect. Once INR is therapeutic then heparin can be stopped - there is actually a period of hypercoaguability when starting warfarin because Protein C and S are inhibited before the other clotting factors |
|
|
Administration of warfarin
|
1. given orally
2. heparin is initiated first, until INR is therapeutic 3. The level of anticoagulation is monitored by INR. In most cases the INR of 2-3 is therapeutics. However, in patients with mechanical heart valves, the range is 2.5-3.5 -- monitor intake of legumes (high in Vitamin K) as this decreases the anticoagulant effect |
|
|
Adverse effects of warfarin
|
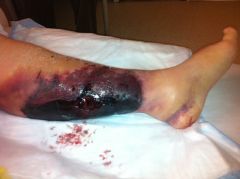
1. hemorrhage
2. skin necrosis is a rare but serious complication. It is causes by a rapid decrease in protein C (a vitamin K dependent inhibitor of factors V and VIII) 3. Teratogenic- AVOID DURING PREGNANCY 4. Should NOT be given to alcoholics or to any patient who is prone to frequent falls because of the increased risk of intracranial bleeds |
|
|
Reversing the effects of warfarin
|
1. Discontinue the warfarin and administer vitamin K
2. The half-life of warfarin is much longer than that of heparin- it takes 5 days to correct the effects of warfarin on stopping the medication. Vitamin K infusion corrects an abnormal PT within 4-10 hours if the patient has normal liver function 3. Giving vitamin K makes it difficult to return the patient to therapeutic INR levels if the anticoagulation is to be continued |
|
|
Clopidogrel- mechanism of action
|
1. Blocks the binding of ADP to a specific ADP receptor, which reduces platelet activation and aggregration
2. increases the bleeding time |
|
|
Clopidogrel- indications for use
|
1. acute coronary syndromes- unstable angina, MI, NSTEMI
2. Pretreatment for patients undergoing PCI 3. After PCI- patients should typically receive 1 year of clopidogrel and asiprin after stent placement 4. Peripheral artery disease |
|
|
Administration of clopidogrel
|
1. given orally
2. for patients with STEMI who will undergo treatment with primary PCI, a loading dose of clopidogrel is associated with better outcomes than pretreatment with placebo. 600 mg dosages seems to have the best risk-benefit ratio. 3. Patients should receive daily clopidogrel after receiving a stent for any indication. The dose is typically 75 mg/day for at least 12 months, with the first 6 months being the most important |
|
|
Adverse effects of clopidogrel
|
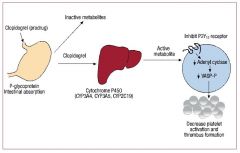
1. bleeding
2. bruising/purpura 3. Some animal models suggest that PPIs interfere with the conversion of clopidogrel to its active metabolite, decreasing its efficacy. For this reason, many people recommend not using clopidogrel with PPIs |
|
|
What are the most common cancers in males (starting with the very most common)
|
1. prostate cancer
2. lung cancer 3. colon cancer |
|
|
The most common cancers in women (very most common first)
|
1. breast cancer
2. lung cancer 3. colon cancer |
|
|
What type of cancer has the highest mortality in both men and women?
|
Lung cancer
|
|
|
What the number on avoidable risk factor for most types of cancer?
|
SMOKING
|
|
|
Risk factors for breast cancer
|
1. prior history of breast cancer
2. age 3. family history of breast cancer 4. female gender 5. anything that increases the number of menstrual cycles (early menarche, late menopause, nulliparity) |
|
|
Approach to a breast mass
|
- breast cancer is rare in women under 35-- so instead think of a fibroadenoma if a young woman presents with a round, movable mass which changes in size over the course of the menstrual cycle
- other breast masses which may occur at any age include cysts (benign if not bloody after aspiration and does not recur) and fibrocystic changes (bilateral breast pain caused by cyclic hormonal stimulation). |
|
|
Ductal carcinoma in situ (DCIS)
|
- premalignant breast cancer- a broad spectrum of diseases that arise from the ductal elements of the breast
- it is most often found on mammogram and mass may or may not be present. - this is a marker for the possible development of invasive ductal carcinoma (typically in teh same breast). - treat with lumpectomy or mastectomy if negative margins cannot be obtained - radiation and systemic chemotherapy are used less often depending on the specific lesion in question |
|
|
Lobular carcinoma in situ of the breast (LCIS)
|
premalignant in situ breast cancer
- arises from the lobular elements of the breast - the lesion is typically found incidentally during biopsy of another breast lesion and a mass is rarely palpable - it leads to an increased risk of breast cancer in either breast. - unfortunately the removal of the lesion does not reduce the risk of progression to invasive cancer - treatment is variable, but may include close observation, selective estrogen modulators and prophylactic bilateral mastectomy |
|
|
What are the two most common types of breast cancer
|
- invasive ductal carcinoma (70% of invasive breast cancers)
- invasive lobular carcinoma |
|
|
presentation of invasive breast cancers
|
- patients may present with a palpable mass, palpable lymph nodes, skin dimpling, nipple retraction or no symptoms at all (found on mammogram)
- |
|
|
Diagnosis of breast cancer
|
- diagnosis must be confirmed with tissue biopsy
- if tumor is the rumor, tissue is the issue, in order for cancer to be the answer! |
|
|
Treatment of breast cancer
|
- treatment typically starts with surgical therapy, though adjuvant chemo is often used
|
|
|
treatment of breast cancers that are < 1cm in size and no lymph node involvement vs treatment of larger lesions
|
- there is no need for either postsurgical chemotherapy or tamoxifen treatment
- large lesions in premenopausal women usually require systemic chemo after surgery - for postmenopausal women with large legions and lymph node involvement, either chemo or tamoxifen is recommended, depending on the status of the estrogen receptor |
|
|
Monoclonal Gammopathy of Undetermined Significance (MGUS)
|
- common in elderly (up to 10% in patients > 75% of age)
- Asymptomatic premalignant clonal plasma cell proliferation - Diagnosis: IgG spike < 3.0 g, less than 10% plasma cells in bone marrow; Bence Jones proteinuria <1 g/24 hours. There should be NO END ORGAN DAMAGE (lytic bone lesions, anemia, hypercalcemia etc) - Fewer than 20% develop multiple myeloma in 10-15 years. However, several studies have shown that almost all patients with multiple myeloma has preceding MGUS - No specific treatment is necessary, just close observation |
|
|
Multiple Myeloma- General Characteristics
1. what is it? 2. epidemiology 3. etiology 4. course |
1. Multiple myeloma is a neoplastic proliferation of a single plasma cell line that produces monoclonal immunoglobulin. This leads to enormous copies of one specific copy of immunoglobulin (usually IgG or IgA type)
2. Incidence is increased after age 50, it is twice as common in African Americans as caucasians 3. The etiology is unclear 4. As the disease advances, bone marrow elements are replaced by malignant plasma cells. Therefore anemia, leucopenia, and thrombocytopenia may be present in advanced disease |
|
|
Clinical features of Multiple Myeloma
1. skeletal 2-3. Blood |
1. Skeletal manifestations- bone pain due to osteolytic lesions, fractures, and vertebral collapse-- occurs especially in the low back or chest (ribs) and jaw (mandible). Pathologic fractures. Loss of height secondary to vertebral collapse
2. Anemia - normocytic normochromic- present in most patients due to bone marrow infiltration and renal failure 3. Recurrent infections secondary to deprivation of normal immunoglobulins, therefore humoral immunity is affected. Most common cause of death - up to 70% of patients die of infection (lung or UTI most common) |
|
|
Clinical features of multiple myeloma
1. renal 2. spinal cord |
1. Renal failure mainly due to myeloma nephrosis- immunoglobulin precipitation in renal tubules leads to tubular casts of Bence Jones proteins. Hypercalcemia also plays role in renal decompensation
2. Cord compression - may occur secondary to plasmacytoma or fractured bone fragment. Though rare, this constitutes a medical emergency. Get MRI of the entire spine and START STEROIDS IMMEDIATELY |
|
|
CRAB mnemonic for signs of multiple myeloma
|
C- Calcium (hypercalcemia)
R- Renal failure A- anemia B- Bone lesions (lytic) |
|
|
If a patient presents with low hemoglobin, high calcium, high serum protein, and poor renal function-- what diagnosis should you suspect?
|
Multiple Myeloma
|
|
|
What are the diagnostic criteria for Multiple Myeloma?
|
- At least 10% abnormal plasma cells in the bone marrow plus one of the following: M-protein in the serum, M-protein in the urine, lytic bone lesions (well-defined radiolucencies on radiographs)- predominantly found in the skull and axial skeleton
|
|
|
What would you expect to see on X-rays of patients with multiple myeloma and what causes this?
|
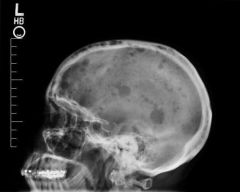
- Radiographs show punched out lytic lesions, osteoporosis, or fractures in 75% of patients with multiple myeloma
- Osteolytic lesions are secondary to the release of osteoclast-activating factor by the neoplastic plasma cells |
|
|
What is the prognosis of multiple myeloma?
|
- the prognosis is poor with a median survival of only 2-4 years with treatment, only a few months without treatment.
- The 5-year survival rate is about 10% |
|
|
Diagnosis of Multiple Myeloma
1. serum and protein electrophoresis 2. plain radiographs 3. bone marrow biopsy 4. Other labs |
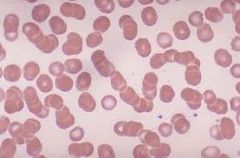
1. Serum and urine protein electrophoresis
a. monoclonal spike due to malignant clone of plasma cells synthesizing a single Ig (usually IgG) called a monoclonal protein (M-protein) b. Serum monoclonal protein is present in 85% of patients, and 75% have a urine monoclonal protein 2. Plain radiographs detect lytic bone lesions 3. Bone marrow biopsy reveals at least 10% abnormal plasma cells 4. Other laboratory findings a. hypercalcemia (due to bone destruction) b. Increased total protein in serum due to paraproteins in blood (hyperglobulinemia) c. Peripheral smear- RBCs are in rouleaux formation, which resembles a stack of poker chips. The hyperglobulinemia causes the RBCs to stick together d. Substantially elevated ESR e. Urine- large amounts of free light chains called Bence Jones protein f. Leukopenia, thrombocytopenia, and anemia may be present, especially in advanced disease g. Elevated creatinine |
|
|
Treatment of multiple myeloma
|
1. In contrast to the premalignant conditions MGUS and smoldering multiple myeloma, patients with full-blown MM require treatment
2. The preferred treatment for multiple myeloma is autologous hematopoietic cell transplantation (HCT), as this has been shown to have higher survival rates when compared to chemotherapy. However, this treatment is usually reserved for younger and relatively asymptomatic patients. If you are considering HCT, you SHOULD NOT start chemotherpay, at this would preclude a patient from having HCT later on 3. Systemic chemotherapy- preferred initial treatment (alkylating agents) for patients who are not transplant candidates 4. Radiation Therapy- if no response to chemotherapy and if the pain is disabling |
|
|
Waldenstroms Macroglobulinemia
|
1. Malignant proliferation of plasmacytoid lymphocytes. These cells produce IgM paraprotein, which is very large and causes hyperviscosity of the blood
2. Diagnosis: IgM > 5 g/dL; Bence Jones proteinuria in 10% of cases, absence of bone lesions 3. Clinical features: fatigue, weight loss, neurologic symptoms, lymphadenopathy, splenomegaly, anemia, abnormal bleeding, and hyperviscosity syndrome (due to elevated IgM). This can lead to retinal vessel dilation with resulting hemorrhage and possible blindness. 4. There is no definitive cure. Use chemotherapy and plasmapheresis for hyperviscosity syndromes. |
|
|
Hodgkin's Lymphoma - general characteristics
1. Epidemiology 2. Lymph node histology |
1. Bimodal age distribution: 15-30 years of age, and > 50 years of age
2. Lymph node histology divides the disease into four subtypes a. lymphocyte predominance (5%)- few Reed Sternberg cells and many B cells b. Nodular sclerosis (70%) -- occurs more frequently in women, bands of collagen envelope pools of Reed Sternberg cells c. mixed cellularity (25%)- large numbers of reen Sternberg cells in a pleomorphic background d. lymphocytic depletion (<1%) - lacking in mix of reactive cells, associated with the worst prognosis - the histologic type does not greatly influence the prognosis of HL unless it is lymphocyte depleted |
|
|
Staging of Hodgkin's Lymphoma
|
- staging is based on physical examination, CT scan (chest, abd and pelvis) and bone marrow biopsy
- Ann Arbor staging system Stage I - confined to single lymph node Stage II - involvement of two or more lymph nodes but confined to same side of the diaphragm Stage III - both sides of the diaphragm involved Stage IV - dissemination of disease to extralymphatic sites Suffixes to stage: A- no symptoms, B- fever, weight loss, night sweats (presence of these constitutional symptoms worsens the prognosis) |
|
|
Clinical features of hodgkin's lymphoma
|
1. Most common symptom is painless lymphadenopathy
2. supraclavicular, cervical, axillary and mediastinal lymph nodes 3. spreads by continuity from one lymph node to adjacent lymph nodes 4. other presentations may or may not be present, including B symptoms (fever, night sweats, weight loss), pruritus, and cough (secondary to mediastinal lymph node involvement) |
|
|
Diagnosis of Hodgkin's lymphoma
|
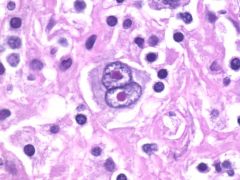
1. lymph node biopsy- The presence of Reed Sternberg cells is required to make the diagnosis.
- neoplastic, large cell with two or more nuclei, look like owl's eye. - usually b-cell phenotype - Reed Sternberg cells may be found in other neoplasms - may be rare in the nodular sclerosis variant 2. Presence of inflammatory cell infiltrates- this distinguishes Hodgkin's lymphoma from NHL. The inflammatory cells present are reactive to Reed Sternberg cells. These include plasma cells, eosinophils, fibroblasts, and T and B lymphocytes 3. CXR and CT scan (chest and abdomen)- to detect lymph node involvement 4. Bone marrow biopsy- to evaluate bone marrow involvement 5. lab findings - leukocytosis, eosinophilia, level of ESR elevation sometimes corresponds with disease activity |
|
|
Treatment of Hodgkin's lymphoma
|
1. Consists mainly of chemotherpy and radiation of the involved field
2. Stages I, II, and IIIa can be treated with radiotherapy alone. However, some physicians advocate the use of chemotherapy in these patients as well 3. Stages IIIB and IV require chemotherapy - Chemo and radiation in combination achieve cure rates of over 70% in Hogkin's lymphoma |
|
|
Non-Hodgkin's lymphoma
1. What is it? 2. how common compared to hodgkin's lymphoma? 3. etiology 4. Epidemiology |
1. NHL is a diverse group of solid tumors which occurs with the malignant transformation and growth of B or T lymphocytes or their precursors in the lymphatic system
a. the type of lymphocyte involved and its level of differentiation determine the course of the disease and its prognosis b. B-cell lymphomas account for 85% of all cases. T-cell lymphomas account for 15% of all cases. c. The disease usually starts in the lymph nodes and may spread to blood and bone marrow. the primary tumor may be found in the GI tract. 2. NHL is twice as common as Hodkgin's lymphoma. At presentation, patients with NHL tend to have more advance disease 3. the etiology is still unknown 4. NHL is the 6th most common cause of cancer-related death in the US. the mean age of onset varies with subtype. There is an increased overall incidence with increasing age |
|
|
Staging of Non-Hodgkin's Lymphoma (NHL)
|
Stage I - single lymph node involved (or one extra lymphatic site)
Stage II- two or more lymph nodes on the same side of the diaphragm (or localized involvement of one lymph node region and a contiguous extralymphatic site) Stage III - lymph node involvement on both sides of the diaphragm Stage IV- disseminated involvement of one or more extralymphatic organs with or without lymph node involvement |
|
|
Risk factors for NHL
|
1. HIV/AIDS
2. Immunosuppression (e.g. organ transplant recipients) 3. History of certain viral infections (e.g. EBV, HTLV-1) 4. History of H. pylori gastritis (risk of primary associated gastric lymphoma) 5. Autoimmune disease -e.g. Hashimoto's thyroiditis or Sjogrens syndrome (risk of mucosa-associated lymphoid tissue - MALT) |
|
|
Classification of Non-Hodgkin's lymphoma (NHL)
|
- there are more than 20 different subtypes of NHL and they are often arranged into unique classification systems. One such classification system stratifies them according to histologic grade: low grade (or indolent), intermediate grade and high grade
|
|
|
Clinical features of Non-hodgkins lymphoma
|
1. Lymphadenopathy- sometimes this is the only manifestation of the disease. Lymph nodes are usually painless, firm, and mobile. Enlargement of lymph nodes is often rapid. Supraclavicular, cervical and axillary nodes are involved most often.
2. B symptoms- less common than in Hodgkin's lymphoma 3. Hepatosplenomegaly, abdominal pain and fullness 4. recurrent infections, symptoms of anemia or thrombocytopenia- due to bone marrow involvement 5. various other findings are possible (SVC syndrome, respiratory involvement, bone pain, skin lesions) |
|
|
Diagnosis of Non-Hodgkin's Lymphoma
|
1. Lymph node biopsy- for definitive diagnosis. Any lymph node > 1 cm present for more than 4 weeks that cannot be attributed to an infection should be biopsied
2. Other tests that may be helpful - CXR- may reveal hilar or mediastinal adenopathy - CT scan (chest, abdomen and pelvis) - to determine the extent of disease spread and patient's response to treatment - Serum LDH and B2 microglobulin are indirect indicators of tumor burden - if alkaline phosphatase is elevated, bone or liver involvement is likely - If liver function tests or bilirubin is elevated then liver involvement is likely - CBC - Serum electrolytes, renal function tests - bone marrow biopsy |
|
|
Prognosis of Non-Hodgkin's Lymphoma
1. Low grade lymphomas 2. Intermediate grade lymphomas 3. High grade lymphomas |
1. Cure is rare. median survival is 5-7 years
2. 50% of patients can be cured with aggressive therapy. Median survival is about 2 years 3. up to 70% can be cured with aggressive therapy. Median survival WITHOUT treatment is a few months |
|
|
Treatment of Non-Hodgkin's lymphoma (NHL)
|
1. This varies depending on the stage and subtype of NHL. There is no always a standard treatment for a given type of NHL.
2. Indolen forms of NHL are not curable, but have a 5-year survival rate of > 75%. These patients are treated in a variety of ways depending on their age: observation, chemo (single agent or combo), radiation 3. Intermediate or high grade NHLs may be curable with aggressive treatments, but if complete remission is not achieved, survival is usually less than 2 years. In general, aggressive forms are treated with multiple regimens of combination chemotherapy (CHOP - cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone) and radiation therapy - Very high dose chemo and BMT is a last resort - rituximab- an anti-CD-20 drug is often used in concert with CHOP (better than CHOP alone) |
|
|
Acute leukemias (2 types)
|
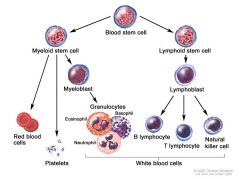
1. Acute Myelogenous Leukemia (AML)
2. Acute lymphocytic Leukemia (ALL) - acute leukemias account for 60% of all leukemias, 25% are CLL and 15% are CML |
|
|
Acute Myelocytic Leukemia (AML)
|
- neoplasm of myelogenous progenitor cells
- AML occurs mostly in adults (accounts for 80% of adult acute leukemias) - risk factors include exposure to radiation, myeloproliferative syndromes, Down's syndrome, and chemotherapy (alkylating agents) - response to therapy is not as favorable with AML as it is with ALL - One important variant of AML is acute promyelocytic leukemia (APL)- this condition is characterized by a t(15,17) and often presents with pancytopenia. Patients may be very sick, so treatment with ALL-TRANS RETINOIC ACIDS (ATRA) should be started without delay with concurrent chemo - may see DIC!!! |
|
|
Acute Lymphoblastic Leukemia (ALL)
|
- ALL is a neoplasm of early lymphocytic precursors. Histology reveals a predominance of lymphoblasts
- ALL is the most common malignancy in children under age 15 in the US - It is the leukemia most responsive to therapy - Poor prognostic indicators are as follows: age < 2 or > 9. WBC >100,000 and/or CNS involvement - presence of any of any of the following is associated with increased risk for CNS involvement: b-cell phenotype, increased LDH, rapid leukemic cell proliferation 2. Many patients with acute leukemias can be cured if treated aggressively. However, the most aggressive forms can be fatal within months |
|
|
Clinical features of acute leukemias
|
1. anemia and associated symptoms
2. increased risk of bacterial infections (due to neutrophilia) - pneumonia, UTI, cellulitis, pharyngitis, esophagitis. Associated with high morbidity and mortality. Potentially life-threatening 3. Abnormal mucosal or cutaneous bleeding (due to thrombocytopenia)- e.g. epistaxis, bleeding at puncture sites, petechiae, ecchymosis 4. splenomegaly, hepatomegaly, lymphadenopathy 5. bone and joint pain (invasion of the periosteum) 6. CNS involvement - diffuse or focal neurologic dysfunction (e.g. meningitis, seizures) 7. testicular involvement (ALL) 8. Anterior mediastinal mass (T-cell ALL) 9. skin nodules (AML) |
|
|
Diagnosis of Acute Leukemias
|
1. Labs
- the WBC count is variable- from 1,000 to 100,000/mm^3. There are a significant number of blast cells (immature cells) in the peripheral blood - anemia - thrombocytopenia - granulocytopenia- put the patient at high risk for infection - electrolyte disturbances- hyperuricemia, hyperkalemia, hyperphosphatemia 2. bone marrow biopsy is required for diagnosis- replacement of marrow by blasts |
|
|
Treatment of acute leukemias
|
1. Treatment of emergencies
- blood cultures, antibiotics for infections - blood transfusion for anemia and platelet transfusion for bleeding, if necessary 2. Aggressive, combination chemotherapy in high doses for several weeks is appropriate to obtain remission (i.e. absent leukemic cells in the bone marrow). Once, remission occurs, maintenance therapy is used for moths or years to prevent recurrence 2. bone marrow transplant |
|
|
Treatment of ALL and prognosis
|
1. More than 75% of children with ALL achieve complete remission (compared with 30-40% of adults). Relapses occur, but usually respond to treatment. With aggressive therapy, survival rates in children can be up 15 years or longer. Up to 50% of patients are cured
|
|
|
Treatment of AML and prognosis
|
AML is more difficult to treat than ALL as it does not respond as well to chemotherapy
- survival rates are considerable lower despite intensive treatment. - BMT gives the best chance of remission or cure |
|
|
Auer Rods
|
clumps of azurophilic granular material that form elongated needles seen in the cytoplasm of leukemic blasts.
- they can be seen in AML M2 and M3 |
|
|
What form of leukemia is DIC most likely a complication?
|
AML M3!!
|
|
|
Chronic Lymphocytic Leukemia (CLL)
|
1. The most common leukemia that occurs after age 50. Most patients with CLL are > 60 years of age. It is the most common leukemia in the Western World
2. The cause is unknown 3. Monoclonal proliferation of lymphocytes that are morphologically mature but functionally defective (i.e. they do not differentiate into antibody-manufacturing plasma cells) 4. In general, this is the least aggressive type of leukemia and CLL patients survive longer than those with acute leukemias or CML. The course is variable, by may follow a prolonged, indolent course. Many patients die of other causes 5. The most recent WHO classification released in 2008 describes CLL as identical to the indolent NHL B-cell neoplasm small lymphocytic leukemia. They are considered the same disease in different stages. |
|
|
Clinical features of chronic lymphocytic leukemia
|
1. Usually asymptomatic at the time of diagnosis. CLL may be discovered on a routine CBC - lymphocytosis
2. Generalized painless lymphadenopathy (lymph nodes are nontender), splenomegaly 3. Frequent respiratory or skin infections due to immune deficiency 4. In more advanced disease: fatigue, weight loss, pallor, skin rashes, easy bruising, bone tenderness and/or abdominal pain |
|
|
Diagnosis of chronic lymphocytic leukemia (CLL)
|
1. Lab findings
a. CBC - WBC 50,000 to 200,000 b. anemia, thrombocytopenia and neutropenia are common c. Peripheral blood smear is diagnostic. Absolute lymphocytosis- almost all of the WBCs are mature, small lymphocytes. Presence of SMUDGE CELLS- "fragile" leukemic cells that are broken when placed on a glass slide d. flow cytometry of the peripheral blood will show clonal population of B cells 2. bone marrow biopsy- presence of infiltrating leukemic cells in bone marrow |
|
|
Treatment of chronic lymphocytic leukemia (CLL)
|
1. Chemotherapy has little effect on overall survival, but is given for symptomatic relief and reduction of infection. Patients are often observed until symptoms develop
2. Prognosis is variable depending on the number of lymph node sites involved and the presence or absence of anemia/thrombocytopenia |
|
|
What cells constitute the myeloid line?
|
Erythrocytes, granulocytes, and platelets
|
|
|
Chronic Myeloid Leukemias- general characteristics
|
1. Neoplastic, clone proliferation of myeloid stem cells
2. Patients are usually older than 40 years of age 3. CML follows an indolent (chronic) course for many years before it transforms to acute leukemia. The end point of the disease course is usually an acute phase (or blast crisis), which is an accelerated phase of blast and promyelocyte production 4. it is associated with a translocation t(9,22)- this philadelphia chromosome. The fusion of the BCR gene on chromosome 22 with the ABL1 gene on chromosome 9 occurs in more than 90% of patients. The abnormal chromosome results in a constituitively active tyrosine kinase protein. Note that patients without the Philadelphia chromosome have shorter survival times and respond more poorly to treatment |
|
|
Clinical features of chronic myelogenous leukemia
|
1. Most patients present in the chronic phase (85%). These patients may be asymptomatic at the time of diagnosis-- disease discovered on routine blood work.
2. constitutional symptoms are also common presenting symptoms- fevers, night sweats, anorexia and weight loss 3. recurrent infections, easy/bruising, symptoms of anemia 4. splenomegaly, hepatomegaly, lymphadenopathy 5. patients may also present in more advanced stages either the accelerate phase or blast crisis |
|
|
Diagnosis of chronic myelogenous leukemia (CML)
|
1. Lab findings - Marked leukocytosis - WBCs from 50,000 to 200,000 with a left shift toward granulocytes
- small number of blasts and promyelocytes - eosinophilia - peripheral smear- leukemic cells in the peripheral blood: myelocytes, metamyelocytes,bands and segmented form - DECREASED LEUKOCYTE ALKALINE PHOSPHATASE ACTIVITY -thrombocytosis - bone marrow biopsy shows leukemic cells |
|
|
What characteristic lab study helps identify CML?
|
decreased leukocyte alkaline phosphatase activity-- and DNA test for 9,22 bcr-abl translocation
|
|
|
Treatment of chronic myelogenous leukemia (CML)
|
1. treatment for CML is one of the great stories of modern medicine. The mechanism of the disease was elucidated and an oral tyrosine kinase inhibitor (imantinib-gleevac) was developed. The drug targets the dysfunctional chimeric protein bcr-abl form by the t(9,22) philadelphia chromosome.
2. unfortunately, patients who present in blast crisis still have very poor outcomes. Imantinib treatment can be attempted and stem cell transplantation is an option |
|
|
Polycythemia Vera- general characteristics
|
1. Malignant clonal proliferation of hematopoietic stem cells leading to excessive erythrocyte production
2. the increase in RBC mass occurs independent of erythropoietin. 3. The median survival with treatment is about 9 to 14 years |
|
|
Clinical features of polycythemia vera
|
1. symptoms are due to hyperviscosity: headache, dizziness, weakness, pruritus, visual impairment, dyspnea
2. Thrombotic phenomena- DVT, CVA, MI and portal vein thrombosis 3. bleeding- GI or GU bleeding, ecchymoses, epistaxis 4. splenomegaly, hepatomegaly 5. HTN |
|
|
Diagnosis of polycythemia vera
|
1. rule out causes of secondary polycythemia (hypoxemia, CO exposure)
2. CBC - elevated RBC count, hemoglobin and hematocrit (usually > 50) 3. serum EPO levels are reduced- compensation 4. elevated B12 level 5. hyperuricemia is common 6. bone marrow biopsy confirms the diagnosis |
|
|
Treatment of polycythemia vera
|
1. repeated phlebotomy to lower HCT
|
|
|
Myelodysplastic syndromes- general characteristics
1, what are they? 2. epidemiology 3. causes 4. classification 5. prognosis |
1. MDS syndromes are a class of acquired clonal blood disorders. They are characterized by ineffective hematopoiesis, with apoptosis of myeloid precursors. The result is pancytopenia, despite a normal or hypercellular bone marrow
2. They occur more commonly in elderly patients, and are slightly more common in men 3. Causes - usually idiopathic, exposure to radiation, immunosuppressive agents, and certain toxins are known risk factors 4. they are classified into subtypes according to the findings on bone marrow biopsy and peripheral smear 5. the prognosis, although variable, is generally poor and the end result is often progression to acute leukemia |
|
|
Clinical features of myelodysplastic syndromes (MDS)
|
1. They are often asymptomatic in the early stages. Pancytopenia may be an incidental finding on routine blood test
2. They may present with manifestations of anemia, thrombocytopenia or neutropenia |
|
|
Diagnosis of myelodysplastic syndrome (MDS)
|
1. bone marrow biopsy typically shows dysplastic marrow cells with blasts or ringed sideroblasts.
2. CBC with peripheral smear shows the following - normal or mildly elevated MCV - low retic count - other abnormalities may include Howell-Jolly bodies, basophilic stippling, nucleated RBCs, hypolobulated neutrophilic nuclei, and large, agranular platelets 3. cytogenic studies often reveal chromosomal abnormalities or mutated oncogenes |
|
|
Treatment of myelodysplastic syndrome (MDS)
|
1. Treatment is mainly supportive
- RBC and platelet transfusions are the mainstays of treatment - Erythropoietin may help reduce the number of blood transfusions necessary - granulocyte colony-stimulating factor can be effective adjunctive treatment - vitamin supplementation, particularly with B6, B12 and folate is important given the large turnover of marrow cells 2. pharmacologic therapies have variable results and include: immunosuppressive agents, chemo, and androgenic steroids 3. BMT is the only potential cure |
|
|
Essential Thrombocythemia
|
- defined as a platelet count > 600,000/mm^3
- a diagnosis of exclusion. Reactive thrombocytosis (due to infection, inflammation, bleeding and so on) and other myeloproliferative disorders must be excluded - It is primarily manifested by thrombosis (e.g. CVA) or paradoxically and less frequently, bleeding (due to defective platelet function). It is a disease with high morbidity and low mortality - other findings many include splenomegaly, pseudohyperkalemia, and elevated bleeding time. Erythromelalgia is burning pain and erythemia of the extremities due to microvascular occlusions - peripheral smear shows hypogranular, abnormally shaped platelets - bone marrow biopsy shows an increased number of megakaryocytes. - treatment usually involves antiplatelet agents such as anagrelide and low-dose aspirin. Hydroxyurea is sometimes used for severe thrombocytosis |
|
|
Diagnostic criteria for polycythemia vera
|
Must have all three major criteria or any two major criteria PLUS any two minor criteria
Major criteria: 1) elevated RBC mass (men > 36 L/kg, women > 32 L/kg, 2) arterial oxygen saturation > 92 % (r/o inc EPO due to lung dz), 3) splenomegaly Minor criteria: 1) thrombocytosis (PLT count > 400 x10^9), 2) leukocytosis > 12 x 10^9, 3) leukocyte alkaline phosphatase > 100 (no fever or infection), 4) serum vitamin B12 > 900 pg/mL |