Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
94 Cards in this Set
- Front
- Back
|
What is hemostasis?
|
The way by which we repair damage to a tube (blood vessel)
|
|
|
In order to form a blood clot (endothelium "bandaid") we have two distinct stages. What are they called?
|
First stage: Primary hemostasis
Second stage: Secondary hemostasis |
|
|
The goal of primary hemostasis is to __________________.
|
form a platelet plug (an adhesion and aggregation of platelets)
|
|
|
The platelet plug in primary hemostasis is ________ (weak/strong).
|
weak
|
|
|
The goal of secondary hemostasis is to __________________.
|
stabilize the previously made platelet plug.
|
|
|
Another word for a thrombus is _____.
|
clot
|
|
|
Primary hemostasis is mediated by interaction between platelets and the endothelial wall. Secondary hemostasis is mediated by ________________.
|
the coagulation cascade
|
|
|
The first step in primary hemostasis is _____________.
|
Transient vasoconstriction of the damaged vessel.
- Mediated by reflex neural stimulation and endothelin release from endothelial cells. |
|
|
The second step in primary hemostasis is _______________.
|
Platelet adhesion to the surface of disrupted vessel
- Von Willebrand factor (vWF) binds exposed subendothelial collagen, - Platelets bind vWF |
|
|
Platelets use __________ to bind Von Willebrands factor.
|
GPIb
|
|
|
vWF is derived from ______________ and _____________.
|
Weibel Palade bodies of endothelial cells and alpha-granules of platelets.
Weibel (W = Willebrand) Palade (P-selectins) |
|
|
The third step in primary hemostasis is _______________.
|
Platelet degranulation & shape change.
- Adhesion induces shape change in platelets and degranulation with release of multiple mediators. |
|
|
ADP promotes upregulation of _____ on platelets. It is essential for _________.
|
GPIIb/IIIa; platelet aggregation
|
|
|
ADP is released from platelet _______________.
|
dense granules
|
|
|
TXA2 is synthesized by platelet _______ and promotes __________.
|
COX; platelet aggregation
|
|
|
The linker molecule that platelets use to aggregate to one another is called _____________.
|
fibrinogen
|
|
|
Disorders of primary hemostasis are usually due to ________________.
|
abnormalities of platelets
|
|
|
Disorders of primary hemostasis are divided into _________ and __________.
|
quantitative; qualitative
|
|
|
The predominant clinical picture of a patient with primary hemostasis disorder is what?
|
Mucosal- and skin bleeding.
|
|
|
Describe symptoms of mucosal bleeding.
|
• Epistaxis
• Hemoptysis • GI bleeding • Hematuria • Menorrhagia (heavy menstrual bleeds) • Petechiae (pin-point bleeding in the skin) • Purpura (slightly larger, 3 mm or greater) • Ecchymoses (measure greater than 1 cm) • Easy bruising |
|
|
One of the feared complications of having a low platelet count is _____________.
|
intracranial bleeding
|
|
|
Patients with qualitative primary hemostasis disorders normally don't get ___________.
|
petechiae
|
|
|
Petechiae is a sign of ___________.
|
Thrombocytopenia (quantitative disorder)
|
|
|
When a patient has disorder of primary hemostasis (mucosal bleeds, skin bleeds) then you're gonna wanna look at lab tests to determine where the problem is. What would be appropriate tests?
|
• PLT count
• Bleeding time (not used much) • Blood smear (we can look at # and size) • Bone marrow biopsy (if PLT is low, could the reason be absent megakaryocytes) |
|
|
Normal PLTs
|
150-400.000 per microliter
|
|
|
Normal bleeding time is
|
2-7 minutes
|
|
|
Chronic ITP is a type _____ hypersensitivity reaction.
|
II
|
|
|
This is the most common cause of thrombocytopenia in children.
|
Acute ITP
|
|
|
This is the most common cause of thrombocytopenia in adults.
|
Chronic ITP
|
|
|
In ITP there are antibodies against ________ on platelets.
|
Gp IIb-IIIa receptors
|
|
|
Acute ITP typically has an abrupt onset _________ weeks after a viral infection.
|
1-3
|
|
|
Acute ITP usually respons _________ (well/poorly) to corticosteroids.
|
well
|
|
|
How is the corticosteroid response in adults with chronic ITP?
|
Adults may show early response, but often relapse
|
|
|
A mother with chronic ITP delivers a newborn. What are your concerns?
|
Anti-PLT IgG can cross the placenta and cause short-lived thrombocytopenia in offspring.
|
|
|
Chronic ITP may be primary or secondary. Mention some causes of secondary ITP.
|
• SLE
• HIV |
|
|
A man with chronic ITP develops spontaneous bleedings from mucosal and skin sites. What could you do to stop bleeding?
|
IV y-globulin temporarily stops serious bleeding (IgG blocks macrophage Fc receptors). It's like throwing a dog a bone.
|
|
|
Chronic ITP is most often seen in __________ (women/men) _________ years of age.
|
women; 20-40
|
|
|
When a patient gets splenectomy (and he/she has ITP) what are you eliminating?
|
• Source of antibodies
• Site of destruction |
|
|
The source of autoantibodies in ITP is ___________ in the __________.
|
plasma cells; spleen
|
|
|
Splenectomy in ITP is only performed in what cases?
|
refractory cases
|
|
|
What is the underlying pathology in microangiopathic hemolytic anemia?
|
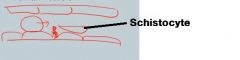
Pathologic formation of platelets in small blood vessels (microangio). RBCs are "sheared" as they cross microthrombi, resulting in hemolytic anemia with schistocytes.
|
|
|
What would you expect cell number-wise in microangiopathic hemolytic anemia.
|
• Low platelets
|
|

What is this cell called?
|
Schistocyte. Sometimes called a helmet (look at the edges).
|
|
|
In what two disorders do we see microangiopathic hemolytic anemia?
|
• TTP
• HUS |
|
|
What is the main problem in TTP?
|
Decreased ADAMTS13, a vWF-cleaving metalloproteinase in endothelial cells. Large, uncleaved multimers lead to abnormal PLT adhesions, resulting in microthrombi.
|
|
|
What is the cause behind decreased ADAMTS13 in TTP?
|
It is usually due to an acquired autoantibody. Could be genetic.
|
|
|
TTP is most commonly seen in _____________.
|
adult females
|
|
|
HUS is due to [...]
|
HUS is due to endothelial damage by drugs or infection.
|
|
|
Infection with a special bug causes HUS. Elaborate.
|
E.coli O157:H7 produces verotoxin which damages endothelial cells. This creates PLT microthrombi, particularly in the kidney, but also in the brain.
|
|
|
HUS is classically seen in __________ (adults/children) who are exposed to _______________.
|
children; undercooked beef
|
|
|
HUS is classically seen in ________________________.
|
children with E. coli O157:H7 dysentery
|
|
|
TTP & HUS are both _________________.
|
microangiopathic hemolytic anemias
|
|
|
What clinical findings are seen in TTP & HUS?
|
• Skin & mucosal bleedings (consumed PLTs)
• Microangiopathic hemolytic uremia • Fever • Renal insufficiency (predominant problem in HUS) • CNS abnormalities (predominant problem in TTP) |
|
|
In both TTP and HUS renal and CNS involvement is seen. What predominates in each disease?
|
In HUS, renal insufficiency is the major problem.
In TTP, CNS abnormalities is the major problem. |
|
|
What laboratory findings would you expect in TTP and HUS?
|
• Thrombocytopenia with increased bleeding time
• PT/PTT are normal • Anemia with schistocytes • Increased megakaryocytes (bone marrow biopsy) |
|
|
True or false: In microangiopathic hemolytic anemia (TTP & HUS) the pathologic microthrombi are stabilized by a fibrin meshwork.
|
False, they are only composed of PLTs and fibrinogen. That's why PT and PTT are classically normal in these disorders.
|
|
|
The treatment of TTP and HUS (particularly TTP, because it is due to an antibody) involves what?
|
• Plasmapheresis
• Corticosteroids |
|
|
HUS may be caused by other infections (other than E.coli). What bugs might be implicated?
|
• Shigella
• Salmonella |
|
|
Bloody diarrhea in HUS are seen in ____% of cases.
|
75%
|
|
|
Mortality rate of HUS is ___ to ___ %.
|
3;5
|
|
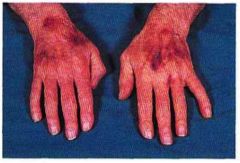
What is this?
|
|
|
|
What is the genetic deficiency in Bernard-Soulier syndrome? What is impaired?
|
Genetic GpIb deficiency. PLT adhesion is impaired.
|
|
|
What would a blood smear show in Bernard-Soulier syndrome?
|
Mild thrombocytopenia; when PLTs don't have GpIb they tend not to live as long. As a consequence of this, platelets are produced which are a little bit bigger than normal, so we see enlarged PLTs.
|
|
|
What is a mnemonic for Bernard-Soulier syndrome to remember that on blood smear we see enlarged PLTs?
|
Bernard-Soulier = B-S = Big-Suckers
|
|
|
What might help you remind you about the pathology in Glanzmann thrombasthenia?
|
Asthenia (Greek: ἀσθένεια, lit. lack of strength but also disease).
The problem is GpIIb-IIIa deficiency (genetic), so that PLTs after activating when interacting with vWF cannot aggregate and link to each other ("weak") because of the absence of the receptor binding to fibrinogen, thus linking cells. |
|
|
Why do we say that aspirin induces a qualitative "problem" with platelets?
|
ASA irreversibly acetylates COX (inactivation). TXA2 cannot be produced, impairing PLT aggregation.
|
|
|
What state induced by failing kidneys may produce a qualitative disorder of platelets? What happens?
|
Uremia.
Both adhesion and aggregation are impaired. |
|
|
What are the qualitative disorders of platelets?
|
Bernard-Soulier syndrome
Glanzmann thrombasthenia Aspirin-induced Uremia |
|
|
Both Bernard-Soulier and Glanzmann are autosomal _______ (recessive/dominant) diseases.
|
recessive. With these diseases you have a lifelong bleeding problem.
|
|
|
When blood is drawn into a clot tube (no anticoagulant is added), a _______ clot is formed. When the
tube is spun down in a centrifuge, the supranate is called ______, which, unlike plasma, is missing ________, ___________, ________, and ___________. |
fibrin; serum; fibrinogen; prothrombin (II), factor V, and factor VIII.
|
|
|
The majority of vitamin K is derived from__________.
|
colonic bacteria; it is synthesized by colonic bacteria
|
|
|
Vitamin K-dependent factors?
|
II, VII, IX, X, protein C, protein S
|
|
|
Vitamin K is activated in the liver by ___________.
|
epoxide reductase
|
|
|
Draw the coagulation pathway
|
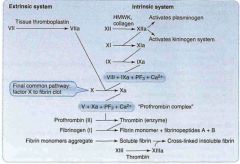
|
|
|
A temporary platelet plug is held together by __________.
A stable platelet plug is held together by _________. |
fibrinogen; fibrin
|
|
|
What is the action of thromboxane A2?
|
(1) TXA2 is a vasoconstrictor, which reduces blood flow.
(2) TXA2 is a platelet aggregator, • Enhances fibrinogen attachment to Gpllb-llla receptors |
|
|
The formation of a temporary platelet plug correlates with what?
|
Bleeding time
|
|
|
Proteins C and S inactivate factors ______ and _______.
|
V; VIII
|
|
|
How is plasmin and proteins C and S similar?
|
Degradation/inactivation of coagulation factors V and VIII
|
|
|
tPA activates _________to release the enzyme ___________.
|
plasminogen; plasmin
|
|
|
Alteplase and reteplase are what?
|
recombinant forms of tPA used in thrombolytic therapy
|
|
|
PTT evaluates __________.
PT evaluates ___________. |
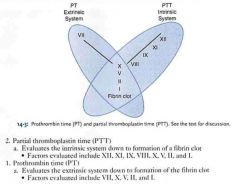
|
|
|
vWF disease is autosomal ___________.
|
dominant
|
|
|
Scurvy makes the blood vessels _________.
|
defective
|
|
|
Normal reference interval for PTT is ______ to _______ seconds.
|
25;40
|
|
|
Whether the patient is anticoagulated with heparin or warfarin, both the PT and PTT are prolonged, because both inhibit factors in the final common pathway. Experience has shown that the PT performs
better in monitoring __________, while the PTT performs better in monitoring _________. |
warfarin; heparin
|
|
|
Normal reference range for PT is _____ to _____ seconds.
|
11; 15
|
|
|
Usual range for INR?
|
2 to 3
|
|
|
Give examples of primary thrombocytosis.
|
• Essential thrombocythemia
• Polycythemia vera |
|
|
Give examples of secondary (reactive) thrombocytosis.
|
• Chronic iron deficiency
• Infections • Splenectomy • Malignancy |
|
|
How do pinpoint bleeds (petechiae) occur?
|
RBCs leak through postcapillary venular gaps in the endothelium.
|
|
|
If bleeding is not severe, treatment is an intramuseular injection of _________.
• Corrects bleeding in a few hours If bleeding is severe, treatment is with __________. |
vitamin K;fresh frozen plasma.
|
|
|
What is the test for vWF?
|
Ristocetin cofactor assay
|
|
|
What is the composition of thrombi in coronary artery thrombosis vs pulmonary thromboembolism and DIC?
|
Coronary artery thrombosis - PLTs held together by fibrin
Pulmonary thromboembolism & DIC - RBCs, PLTs, WBCs held together by fibrin. |