Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
35 Cards in this Set
- Front
- Back
|
Follicular carcinoma
|
Follicular carcinoma
2nd most common form of THYROID cancer (5-15% of cases) M:F=1:3 and peak incidence at 40-60 years Two forms: 1. adenoma-like form 2. invasive form Mutations in PI-3K/AKT pathway in ~33% (RAS gain of function, PTEN loss) & PAX-8 - PPAR∂1 translocations in 30-50% Often forms follicles and occasionally the cells are either clear or oxyphilic (Hurthle cell) Metastasis frequently hematogenous (unlike the lymphatic metastasis of the papillary thyroid carcinoma.) - Mets of follicular carcinoma may be found in the lung, bones, or sometimes in the CNS. Radioiodine therapy- Why does this work? The tumor cells concentrate iodine. Therefore, Radioiodine therapy can cause the local release of radiation to the neoplastic epithelium. 10 year survival is ~90%, but 80% at 20 years and 70% at 30 years so late mortality may occur |
|
|
Medullary carcinoma
|
Medullary carcinoma
~5% of THYROID cancer May be component of MEN 2a syndrome (also has pheochromocytoma and parathyroid hyperplasia) Pheochromocytoma is a rare tumor of the adrenal gland that causes too much release of epinephrine and norepinephrine -- hormones that regulate heart rate and blood pressure. May be component of MEN 2b (pheochromocytoma and mucosal neuromas; also marfanoid) MEN 2a & MEN 2b inherited as autosomal dominants with germline point mutations in the RET proto-oncogene on chromosome 10 causing chronic activation of RET in the parafollicular cells More common for medullary carcinoma to occur sporadically (70% of cases); sporadic tumors peak in 50s; MEN tumors may occur in children Derived from C-cells (parafollicular, calcitonin producing cells) of thyroid and calcitonin provides marker to detect and follow patients May also secrete ACTH, VIP, serotonin and other substances |
|
|
MEN1
|
MEN1 (multiple endocrine neoplasia) is associated with Pit. tumors
|
|
|
MEN 2a
|
MEN 2a (multiple endocrine neoplasia) and MEN type 2 is associated with:
1. medullary carcinoma of the thyroid 2. parathyroid hyperplasia 3. pheochromocytoma (a tumor of the adrenal gland) However, it's more common for medullary carcinoma to occur sporadically (70% of cases). |
|
|
MEN 2b
|
MEN 2b (multiple endocrine neoplasia) aka MEN type 3 is associated with:
1. medullary carcinoma of the thyroid 2. mucosal neuromas 3. pheochromocytoma (a tumor of the adrenal gland) 4. Marfan syndrome - like habitus However, it's more common for medullary carcinoma to occur sporadically (70% of cases). |
|
|
Medullary carcinoma is derived from __________.
|
Medullary carcinoma is derived from C-cells (parafollicular, calcitonin producing cells) of thyroid and calcitonin provides marker to detect and follow patients
|
|
|
Medullary carcinomas secrete what substances?
|
1. calcitonin
2. ACTH - may cause paraneoplastic Cushing's syndrome 3. VIP 4. serotonin and other substances |
|
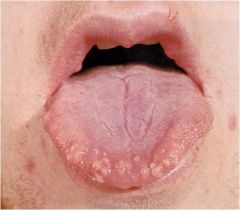
What is this? What causes this?
|
MEN 2B – Mucosal neuromas
|
|
|
Grossly, how do sporadic tumors differ from tumors caused by genetics?
|
Sporadic tumors tend to be large and fleshy at presentation; familial cases resected prophylactically are small and multifocal
|
|
|
What is the most aggressive form of medullary carcinoma?
|
MEN-2b associated medullary carcinoma is more aggressive than other forms such as MEN-2a or the spontaneous form.
|
|
|
Anaplastic carcinoma
|
Anaplastic carcinoma of the thyroid:
Several morphologies: giant cell, spindle cell and combinations Many patients have prior or concurrent differentiated thyroid carcinoma (follicular or papillary), supporting that anaplastic thyroid carcinoma arise from “dedifferentiation” Usually rapid growth in older patient (mean, 65 years) and poor prognosis Patients present with neck mass or airway obstruction secondary to tracheal invasion. Virtually no survivors |
|
|
Anaplastic carcinoma has several _____________.
|
Anaplastic carcinoma has several morphologies: giant cell, spindle cell and combinations
|
|
|
Anaplastic carcinoma arises from ____________.
|
Anaplastic carcinoma arises from dedifferentiation.
Many patients have prior or concurrent DIFFERENTIATED thyroid carcinoma (follicular or papillary), supporting that anaplastic thyroid carcinoma arise from “dedifferentiation” |
|
|
How does a person with anaplastic carcinoma present?
|
Patients present with neck mass or airway obstruction secondary to tracheal invasion. The CA grows though the cartilage of the trachea!!!
Usually rapid growth in older patient (mean, 65 years) and poor prognosis Virtually no survivors |
|
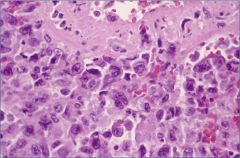
What is this?
|
Anaplastic carcinoma – giant cell variant
|
|
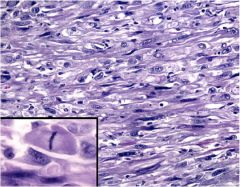
What is this?
|
Anaplastic carcinoma of thyroid – spindle cell type
|
|
|
Parathyroids: embryology & development
|
The Parathyroids develop from the 3rd & 4th pharyngeal pouches – 3rd pouch gives rise to inferior glands & 4th superior
6% of population has > 4 parathyroids; 10% have only 2 or 3 Variable locations: usually upper at level of middle 1/3 of thyroid & lower at inferior poles of thyroid, but sometimes intrathyroidal or mediastinal Fat & oxyphil cells develop at puberty and increase into young adulthood - In childhood, the parathyroids are more cellular with chief cells, but these chief cells undergo oxyphil metaplasia after childhood. |
|
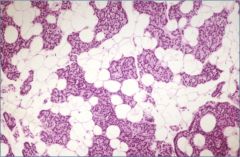
What is this?
|
Normal adult parathyroid gland – note ~ equal amounts of parathyroid cells & fat (50%:50%)
|
|
|
Hypoparathyroidism
|
Causes:
1. Iatrogenic (most common, usually following complete thyroidectomy) 2. Agenesis: (associated with thymic agenesis [DiGeorge syndrome] & aortic arch abnormalties) 3. Autoimmune destruction: associated with PA, Addison’s, mucocutaneous candidiasis & ectodermal dystrophy (APS 1) or isolated parathyroid destruction (Abs vs Ca++ binding receptor) 4. Genetic: Either autosomal recessive (parathyroid maldevelopment) or autosomal dominant (pro-PTH mutation; or calcium receptor mutation causing activation and PTH suppression - the receptor thinks that Calcium is present when its not). Features: low calcium, high PO4, increased bone density, cataracts, calcification in basal ganglia & soft tissues, tetany, Q-T lengthened, psychiatric problems |
|
|
What do the parathyroid glands do?
|
The major function of the parathyroid glands is to maintain the body's calcium level within a very narrow range, so that the nervous and muscular systems can function properly.
When blood calcium levels drop below a certain point, calcium-sensing receptors in the parathyroid gland are activated to release hormone into the blood. Parathyroid hormone (PTH, also known as parathormone) is a small protein that takes part in the control of calcium and phosphate homeostasis, as well as bone physiology. Parathyroid hormone has effects antagonistic to those of calcitonin. PTH increases blood calcium levels by stimulating osteoclasts to break down bone and release calcium. PTH also increases gastrointestinal calcium absorption by activating vitamin D, and promotes calcium uptake by the kidneys. PTH affects the perception of well being and absence of PTH can be associated with feeling of fatigue and anxiety. |
|
|
What are the features of Hypoparathyroidism?
|
Features: low calcium, high PO4, increased bone density, cataracts, calcification in basal ganglia & soft tissues, tetany (involuntary contraction of muscles), Q-T lengthened on EKG, psychiatric problems
|
|
|
Primary hyperparathyroidism
|
Primary hyperparathyroidism
M:F = 1:4 Presentation: increases calcium, increases renal stones, etc. (increased calcium is assymptomatic, increased renal stones are symptomatic.) Usually single adenoma, of inferior glands, with pleomorphism but not mitoses Double adenomas are rare Unilateral neck exploration if one large and one small gland found Sometimes need to do thyroidectomy or mediastinal exploration Asymmetric hyperplasia may mimic adenoma Several genetic changes: MEN1 inactivation (even in sporadic), cyclin D1 gene inversions (sporadic), RET activation (MEN 2 cases), inactivation of calcium sensing receptor (familial hypocalciuric hypercalcemia) |
|
|
What are the 4 top clinical causes of hyperparathyroidism?
|
4 top clinical causes of hyperparathyroidism:
1. Cancer - metastasis to bone on ectopic bone degradation 2. Hypoparathyroidism 3. Vitamin D excess (its major role is to increase the flow of calcium into the bloodstream, by promoting absorption of calcium and phosphorus from food in the intestines, and reabsorption of calcium in the kidneys; enabling normal mineralization of bone and preventing hypocalcemic tetany. - If there's enough calcium available in the blood, because of Vit D, then bone doesn't have to be broken down. This is ow Vit D keeps bones strong) 4. Hyperthyroidism (ex. graves disease) |
|
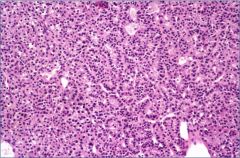
This is the Gland of patient with Primary hyperparathyroidism: What's the cause?
|
You don't know.
This is definitely NOT a normal parathyroid because of the hypercellularity. A normal parathyroid should be 50% fat and 50% oxyphil cells in an adult. But the cause can either be parathyroid adenoma or parathyroid hyperplasia. So what do you do? Remove a 2nd gland from the patient. IF the 2nd gland has a normal fat to cell ratio, then the diagnosis is parathyroid adenoma and you've already treated the patient because the 1st gland was removed. IF the 2nd gland was also hypercellular then you can diagnose parathyroid hyperplasia. In this case, you have to go back in a remove all parathyroids excepts for half of a gland. (You remove 3.5 parathyroid glands) |
|
|
Secondary hyperparathyroidism
|
Secondary hyperparathyroidism = the physiologic development of parathyroid hyperplasia secondary to hypocalcemia.
What causes the hypocalcemia? Usually renal failure with hypocalcemia ( decreased 1,25 di-OH vit D3 and increased PO4 are important factors related to hypocalcemia in chronic renal failure) - People with renal failure don't efficiently hydroxylate their Vit D to make it into the active form. They also don't excrete phosphate very well. Therefore phosphate increases and calcium decreases. Secondary parathyroid hyperplasia develops because of chronic Ca++ decrease (this affects all of the parathyroid glands) |
|
|
“Tertiary hyperparathyroidism”
|
“Tertiary hyperparathyroidism”
Development of autonomy = “tertiary hyperparathyroidism” - even after hypocalcemia has been treated, an rouge parathyroid gland doesn't care and it just keeps on secreting PTH. |
|
|
Parathyroid carcinoma
|
Parathyroid carcinoma
Rare disease May cause extreme increase in Ca++ Often difficult histologic diagnosis - best evidence for cancer may be invasion of the surrounding soft tissue at surgery 30-40% 5 year survival; poor prognosis if local recurrence develops within the first 2 years |
|
|
Hyperparathyroid effects
|
Hyperparathyroid effects:
1. Renal stones -----> pyelonephritis (kidney infections) 2. Osteitis fibrosa cystica, with “brown” tumors secondary to net osteoclastic activity 3. Corneal calcification (band keratopathy) 4. Peptic ulcers - because of an increase in the release of acid from the stomach because of PTH 5. Pancreatitis 6. Cardiac valve calcifications |
|
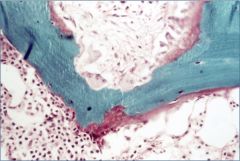
What is this?
|
Hyperparathyroidism – osteitis fibrosa
Osteitis fibrosa cystica, with “brown” tumors secondary to net osteoclastic activity |
|
|
Explain how the Adrenal medulla and the Sympathetic paraganglionic system are related.
|
Both the Adrenal medulla and the sympathetic paraganglionic cells are derived from the neural crest. The adrenal medulla is composed of chromaffin cells that release catecholamines (epinephrine and norepinephrine) and the paraganglia are nodular, pea sized structures lying along or within nerves that contain clusters or cord of chromafin cells that also secrete catecholamines.
|
|
|
Adrenal medulla/Sympathetic paraganglionic system
|
Adrenal medulla/Sympathetic paraganglionic system
Neural crest origin Synthesize catecholamines (epinephrine & norepinephrine) Single largest collection = adrenal medulla but other sites include neck, mediastinum, retroperitoneum & viscera Sympathetic paraganglionic cells sometimes called “chromaffin” cells |
|
|
Pheochromocytoma
|
Pheochromocytoma = a tumor within the adrenal MEDULLA
Derived from chromaffin cells (paraganglionic tissue) “10% tumor” - This means that: - 10% of Pheochromocytomas are extra-adrenal and are called paragangliomas - 10% of Pheochromocytomas are malignant -10% of Pheochromocytomas are bilateral [but higher if MEN or other] -10% of Pheochromocytomas are non-hypertensive and 10% of Pheochromocytomas occur during childhood. May be component of MEN 2/3, von Hippel Lindau (vascular tumors, RCC, cerebellar hemangioblastoma), NF1, or isolated familial Episodic or sustained hypertension caused by catecholamine release Extra-adrenal (paraganglioma) associated with greater nor-epinephrine Path: brown tumor with large cells having granular cytoplasm arranged in “zellballen” Malignancy difficult to predict short of metastasis |
|
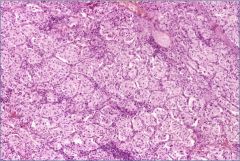
What is this?
|
Pheochromocytoma – note “zellballen”
“Zellballen” = cell balls = tight clusters of tumor cells surrounded by blood vessels or fibrous tissue. |
|
|
Neuroblastoma
|
Neuroblastoma = a tumor mostly in children, derived from residual sympathetic neuroblastic cells.
80% in < 5 year olds; median is 1.5 years Some cases associated with NF; also familial cases (germline ALK mutation) 65% are intra-abdominal (in the adrenal gland) Prognosis depends on : 1. age (better if <1.5 years), 2. site (better if mediastinal), 3. stage (better if early, except for IVS), IV-S = Stage 4 Special = a types of neuroblatoma confined to the bones, skin and liver. Although this is a stage 4 tumor, patients do very well. 4. genetic features (better if NO N-myc amplification, 17q gain or 1p loss) and 5. histology (better if differentiation & Schwannian stroma) 90% secrete catecholamines, but rarely cause hypertension Path: brown/gray, necrosis, calcification; small dark neuroblastic cells in neuropil with rosettes, sometimes ganglionic cell differentiation Prognosis: 40-50% 5 year survival overall |
|
|
Neuroblastoma Progression
|
1. immature embryonic cells resembling neoplasm
2. differentiating/ ganglionic neuroblastoma 3. Ganglioneuroma (benign and patients are fine) |