Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
71 Cards in this Set
- Front
- Back
|
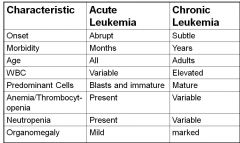
Acute vs Chronic Leukemia
|
|
|
|
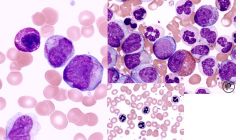
AML smear findings
|
-blasts
-auer rods (faggot cells) --> only monoblast, promyelocyte, myleoblast -anemia -giant plt -nRBCs -pseudo HYPO seg (psuedo pelger huet) or HYPERseg neutrophils |
|
|
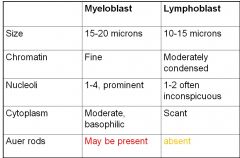
myleoblast vs lymphoblast
|
|
|
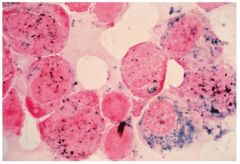
What is this showing? what is its significance
|
-MPO --myeloperoxidase cytochemical stain
-stains primary granules that contain peroxidase -strong ++++ = promyelocytes, myelocytes, myeloblast, metamyelocyte, band, segs - neg stain = differentiates ALL (-) from AML (+) |
|

what is this showing? what is its significance
|
-Sudan black B cytochem stain
-stains phospholipids and other intracellular lipids found in prim and sec grans of myeloid cells - neg in lymphocytes -AML (+), ALL (-) -strong ++ = myeloblasts |
|
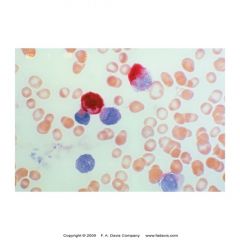
what is this showing? what is its significance
|
-specific esterase cytochem stain
-aka CAE stain -prim gran of myeloid cells -strong++++ = myeloblasts -(+) = seg, baso, mast -neg = eos, mono, lymphocytes *specific for only few in myeloid series |
|
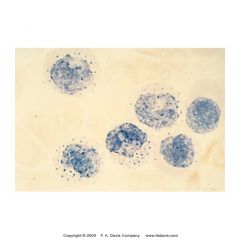
what is this showing? what is its significance
|
-Non specific esterase (NSE) cytochem stain
-strong +++++ = monoblasts -(+) = monocyte, histocyte, macrophage, megakaryocyte -neg = granulocytes -used to ID acute monoblastic and myelomonoblastic leukemia |
|

what is this showing? what is its significance
|
-terminal deoxynucleotidyl transferase (TdT) cytochem stain
-intranuclear enzyme found in stem cell and immature lymphoid cells (B or T) w/in BM -90% lymphoblastic leuk (B or T) -5-10% myeloblastic leuk -30% of blast crisis phase of CML - (+) = lymphoblasts -not commonly done -ALL |
|
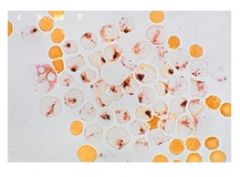
what is this showing? what is its significance
|
Acid phosphatase cytochem stain
-stains t lymphoblasts |
|
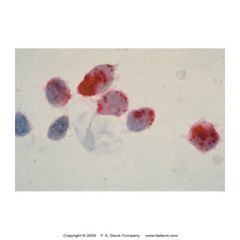
what is this showing? what is its significance
|
-Tartate-resistant acid phosphatase (TRAP) cytochem stain
-stains B lymphoctes in hairy cell leuk (reistant to stain) |
|

what is this showing? what is its significance
|
Periodic acid -schiff (PAS) cytochem stain
-stains glycoproteins and glycogen -ALL (left) - "block staining" in lymphoblasts (around the edges) -erythroleukemia (right) - intense granular pattern in erythroblasts |
|
|
Acute Leukemia CD markers
B, t, erythoid, mega, stem cell |
B cell - 10, 19, 20, 22
T cell - 2-5, 7, 8 erythroid - 13, 33 megakar - 41, 61 stem cell - 34 |
|
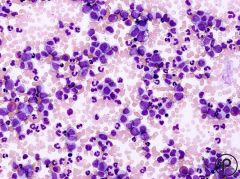
what does this show?
|
CML chronic phase
- cells of all maturity lvls |
|
|
leukamoid rxn vs leukemia
|

-LAP (NAP) stain
-increased in Leukemoid reaction and MPDs except CML -decreased to normal in CML -Eos & Basos -CML has increased numbers of eos and basos -Leukemoid reaction will have decreased to normal eos and basos -Toxic Morphologic Changes -CML without infection or treatment does not exhibit toxic changes -Leukemoid reaction can show toxic changes (e.g. toxic granulation, dohle bodies etc.) |
|
|
CML
|
Chronic myelogenous leukemia
-Severe leukocytosis -Entire spectrum of myeloid cell development** -Eosinophils and basophils are increased -Thrombocytosis present in chronic phase -Blasts increased in acute phase -Bone marrow shows hyperplasia and may be fibrotic -M:E ratio can be as high as 25:1 -WBC > 100 x 109/L -Low leukocyte alkaline phosphatase (LAP) score (<10)** -Normocytic, normochromic anemia** -Increased LDH, uric acid, B12 due to increased catabolism of cellular nuclei -Ph1*** |
|
|
PV - polycythemia vera
|
-RBCs are significantly more sensitive to EPO than usual --> increased RBC#, Hgb and Hct.
-Absolute increase in RBC mass -Mutations likely (JAK 2) -Increased RBC, Hgb and Hct -blood EPO lvls LOW RX: Therapeutic phlebotomy |
|
|
secondary erthrocytosis
|
-NOT autonomous erythropoiesis.
-Due to increased EPO in all cases. -Due to Compensatory increase in EPO R/T hypoxia (eg. respiratory dz., increased altitude) -Pathological secretion of EPO (eg. renal tumor) -Defective oxygen transport (eg. Smoking and CO, environmental pollution) |
|
|
Relative erythrocytosis
|
-Increased Hct but normal RCM
-Decreased plasma volume -Ex. dehydration -Ex. Stress erythrocytosis (Gaisbock’s Syndrome) Hct increases proportionally to hypertension. |
|
|
Essential thrombocythemia
|
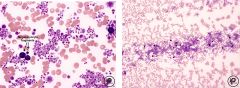
-PLT > 600 x 10 9/L (can be > 1 million/uL!)**
-Usually normal PLT morphology, but can see increased PDW and plt anisocytosis -Megakaryocytic hyperplasia in BM, but can also see increases in other cell lines -Absence of identifiable causes of reactive thrombocytosis -NO Philadelphia chromosome -Hgb no higher than 13 g/dL or normal RCM -NO sig marrow fibrosis (no tear drops) -Presence of stainable iron in BM |
|
|
myelofibrosis with myeloid metaplasia (MMM)
|

-Normochromic normocytic anemia
-Leukoerythroblastosis (incr.NRBC, immature whites and PLT abnormalities) -Many nRBC’s -Tear drops (due to splenic and marrow fibrosis) -Micromegakaryocytes (Dwarf) with cytoplasmic blebs -Giant platelets -Note that PLT and WBC can be increased at first, but then decrease due to fibrotic BM -Prolonged PT, PTT and occult DIC -Hgb < 10 mg/dL -BM is frequently a dry tap -Chromosome abnormalities are common, but NO Philadelphia Chromosome! |
|
|
Chronic lympocytic leuk (CLL)
|
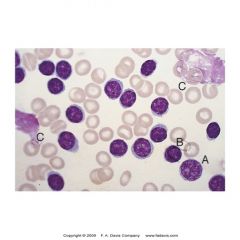
-clonal proliferation of B lymphocytes
-abnormalities on chromosomes 11, 12, and 13, especially trisomy 12 -Malignant lymphocytes have B cell markers CD15, 19, 20, 22 and CD 5 (usually only found on T cells) -Malignant Lymphs exhibit low levels of surface immunoglobulins (SIg) -Small lymphocytes accumulate in the spleen, lymph nodes, and bone marrow and can spill out into peripheral circulation -White counts are exaggerated with many >100,000/L -M:E ratio 10-20:1 -shows exclusively small (adult-looking) lymphocytes. -Smudge cells may be present AKA basket cells -Pieces of lymphocyte chromatin on smear -Lymphocytes are fragile -Albumin smear needed to keep cells intact for counting |
|
|
Hairy cell leukemia
|
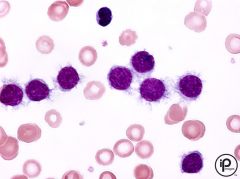
-Hairy cells on peripheral smear
-TRAP positive cells -CD19, 20, 22, 11c, 25, 103 -Rare B cell malignancy -Fragile mononuclear cell |
|
|
Sezary syndrome
|
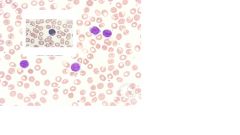
-leukemic phase of this T-cell lymphoma.
-T-cell lymphoma with mycosis fungoides = cutaneous manifestation -Skin biopsies reveal an infiltration of lymphocytes -Spleen, bone marrow, and lymph nodes may become affected -Sezary cells in peripheral blood -Large cells (8 20 m) -Ovoid, convoluted, cerebriform nucleus -May be mistaken for monocytes -pathognomonic of cutaneous T-cell lymphoma -CD2,3,4,5**** |
|
|
Prolymphocytic leuk
|
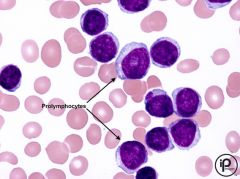
-CLL converts to PLL
-majority of circ = prolymphocyte --> not maturing Prolymphocytes on peripheral smear Strong CD20 and Sig activity Also pos for CD19 |
|
|
Hodgkin's lymphoma
|

-most common lymphomas in young males 12-40 &
-Also seen in people over 50 -single cervical lymph node becomes firm to the touch and does not disappear -People exposed to EBV or environmental hazards are at increased risk. -Diagnosis based on lymph node biopsy ***Presence of Reed-Sternberg cell -Large, Multinucleated, “owl’s eye” -May involve liver, spleen, and bone marrow MOST curable!!!!!! |
|
|
Non Hodgkin's lymphoma
|
-May present as painless cervical lymph node involvement
-Lymphoma cells may be seen in peripheral smear -Diagnosis based on histological types of lymphocytic cells -Treatment includes radiation and chemotherapy -Relapse is frequent -Disease may spread: Gastrointestinal, Respiratory, Skin, Liver, Spleen |
|
|
Multiple Myeloma
|
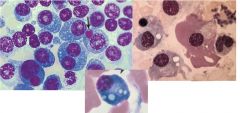
-Disorder of plasma cells: accum of plasma cells in BM and other locations
-older age -environmental and occupational factors -chromosome abnormalities, esp 13 3 PW: 1. Accel of plasma cells in the BM by cytokine IL-6 -May dev colorless inclusions=Russell bodies -Flame cells may be seen in IgA myeloma (Deep pink cytoplasm) -Overcrowding of BM with plasma cells --- May seed to other areas in the body 2. Activation of bone resorption (osteoclasts) -Increased osteoclast activity -bone loss -Serum Ca inc --> kidney failure or stones 3. Prod of an abnorm monoclonal protein -Monoclonal gammopathy -Serum Free Light Chains (Serum beta microglobulin ) =light chains unattached to heavy chains -Rouleaux therefore inc ESR -Bence-Jones protein found in urine (free lt chain) |
|
|
Waldenstrom’s Macroglobulinemia
|
-Overproduction of IgM caused by abnormal B lymphocytes
-Increased IgM interferes with coagulation factors by coating platelets and impeding their function -Hyperviscosity synodrome – slow flowing blood may lead to CNS symptoms ex. Vision disturbances, dizziness. -Peripheral smear=rouleaux and plasmacytoid lymphocytes ==inc ESR -Cryoglobulins may be found in some patients -lead to Raynaud’s phenomenon-cold hands feet -Causes bleeding |
|
|
Myelodysplastic Syndromes (MDS)
|
-Clonal hematologic malignancies
-Hypercellular BM (hyperplasia) -Hematopoiesis is ineffective -Intramedullary death of blood cell precursors via apoptosis -Peripheral blood cytopenias of one or more cell line -Dysplastic blood cells -Refractory macrocytic anemia– multiple transfusions lead to iron overload – may necessitate Fe chelating therapy********** -Propensity to transform into acute leukemia (30-40% of all MDS) |
|
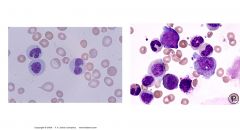
MPD vs MDS
|
MPD (right): Leukocytosis, erythrocytosis, thrombocytosis; all levels of maturation noted (CML in peripheral smear looks like a bone marrow smear – all lines present)
MDS (left) : Hypercellular bone marrow, BUT Cytopenias of one or more cell line on peripheral smear; Ineffective hematopoiesis; intramedullary death of precursor cells; Cells look weird and huge and blasts may be present |
|
|
MDS
Signs and Symptoms |
-Hypercellular marrow
-Macrocytic anemia (called “megaloblastiod” but not responsive to B12 or folate supplementation) -Weakness/Infection -Easy bruisability -Ringed sideroblasts -BM cytoplasmic changes: Vacuolization, Poor gran -BM nuclear changes: Multinuclearity, Disintegration of the nucleus (pyknotic) , N:C asynchrony as in megaloblastic anemia, Nuclear bridging between cells -Dysgranulopoietic changes: hypo/hyper gran, hypo/hyper seg, Pseudo-Pelger-Huet, Degenerating neutrophils -Dyserythropoietic changes: Bizarre nuclear changes, Multinuclear, Nuclear bridging, Macrocytes, Ringed sideroblasts, dimorphism -Dysthrombopoietic changes: Platelet anisocytosis Micromegakaryocytes, Abnorm granularity of platelets, Giant platelets |
|
|
Refractory anemia (RA)
|
Erythroid hyperplasia in BM with dysplasia
Myeloblasts <1% in blood, <5% in BM |
|
|
Refractory anemia with ringed sideroblasts (RARS)
|
Refractory anemia in which 15% or more of red cell precursors are ringed sideroblasts
<5% myeloblasts in BM |
|
|
Refractory anemia with multilineage dysplasia
|
Bone marrow failure with two or more myeloid cell lines affected
Multiple chromosomal abnormalities <1% blasts in peripheral blood |
|
|
Refractory anemia with excess blasts (RAEB)
|
Shows anemia, thrombocytopenia and neutropenia
Hypercellular marrow in most cases 5-19% myeloblasts in BM |
|
|
Myelodysplastic syndrome unclassifiable
|
Features of MDS but not a clear classification
Neutropenia and thrombocytopenia are common |
|
|
Transferrin
|
– a glycoprotein made in the liver that transports iron in the serum
|
|
|
Ferritin –
|
The “storage form” of iron found in tissues, mostly the RES (endothelial cells of liver, spleen and bone marrow). Serum values can be measured.
|
|
|
Porphyrin
|
– organic pigmented molecule that tends to bind with metals
|
|
|
Protoporphyrin –
|
the iron-containing red porphyrin that forms hemoglobin and myoglobin
|
|
|
Red Cell Composition
|
-Heme
4 iron atoms Fe2+ Iron surrounded by protoporphyrin ring -Globin 2 pairs of globin chains made up of amino acids Alpha and beta chains -2,3-Diphosphoglycerate (2,3-DPG) Related to oxygen affinity Produced in the Embden-Meyerhof pathway via Luerbering-Rapaport shunt |
|
|
Struture Hgb
|
4 globin chains
4 heme groups, each consisting of a protoporphyrin ring plus ferrous iron (Fe2+) |
|
|
Protoporphyrin synthesis
|
Delta aminolevulinic acid + EPO+ vit B6 = porphyrinogens
Porphyrinogens - oxidized to protoporhyrins |
|
|
globin synth
|
Genes on chromosomes 16 and 11 code for Hb.
|
|
|
Embryonic Hgb
|
gower
portland epsilon, zeta |
|
|
fetal hgb
|
Hemoglobin F (a2, g2)
alpha, gamma |
|
|
Adult hgb
|
Hemoglobin A (a2, b2) 95 – 97%
Hemoglobin A2 (a2, d2) 1 – 3% Hemoglobin F (a2, g2) 1-2% |
|
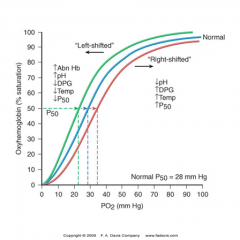
oxygen dissociation curve
|
Shift to right
-More likely to release oxygen to tissue -Lower affinity -Anemia -Acidosis -Increased 2,3-DPG -increased temperature Shift to left -Less likely to release oxygen to tissue -Higher affinity -Decreased body temperature -Abnormal hemoglobin -Alkalosis -Decreased 2,3-DPG -the presence of methemoglobin or carboxyhemoglobin -increased Hb F -multiple transfusions with DPG-depleted stored blood. |
|
|
Carboxyhemoglobin
|
Hb is bound with CO
CO has 200 X the affinity as oxygen for Hb Result is that oxygen can’t bind and tissues become hypoxic. Smoking, accidental exposure to CO emissions, suicide Rx is hyperbaric oxygen administration LEFT shift |
|
|
Methemoglobin
|
Iron in the heme is oxidized to the ferric Fe3+ state, which cannot bind oxygen
Normally only 1% of circulating hemoglobin Results in inability to deliver oxygen to tissues AND promotes increased O2 binding affinity to any normal Hb present, thus the left shift in the oxygen dissociation curve. Main Symptom is cyanosis Happens during oxidative stress, oxidizing drugs, or certain enzyme deficiencies within RBC’s Rx is reducing drugs (ex. New Methylene Blue) LEFT shift |
|
|
Sulfhemoglobin
|
Sulfur irreversibly binds to Hb and no oxygen can bind.
Happens in chronic constipation, sulfur-containing drugs No Rx. RBC’s must be removed from circulation. |
|
|
extravascular hemolysis
|
90%
-destroyed by the RES system = spleen Globin - amino acids - recycled Heme - iron - via transferrin to bone marrow Heme - part of protoporphyrin ring- CO exhaled Heme - other part of the protoporphyrin ring - biliverdin-unconjugated biliruben (via albumin)- liver-conjugated biliruben (some reabsorbed to blood)-bile and intestines -urobilinogen (some reabsorbed to blood)-feces |
|
|
intravascular hemolysis
|
10%
-RBC ruptures in general circulation -Hemoglobin dimers are carried via haptoglobin to the liver -Hb destruction proceeds as per Extravascular hemolysis |
|
|
hemolytic lab findings
|
LOW Hemoglobin, hematocrit, and red cell count
HIGH Serum (unconjugated) bilirubin LOW Serum haptoglobin HIGH Reticulocyte count HIGH Lactate dehydrogenase (LDH) (used in Embden-Meyerhoff pathway: pyruvate to Lactate) Hemoglobinemia Hemoglobinuria Hepatosplenomegaly may be seen |
|
|
Anemia
|
Normocytic (MCV = 80-100)
Macrocytic (MCV > 100) Microcytic (MCV < 80 ) Tip: A normal RBC is the same size or a little smaller than a normal lymphocyte nucleus Normochromic (MCHC = 32-36 %) Hypochromic (MCHC < 32 %) |
|
|
3 pw to anemia
|
Decreased Production of RBCs/Hgb : IDA, AOI, megaloblastic a., aplastic a., leukemia, MPD (myeloproliferative disorders), sideroblastic a.
Increased Destruction of RBCs/Hgb: Hemolytic anemias (immune and non-immune), intracorpuscular and extracorpuscluar defects Loss of RBCs w/o replacement : hemorrhage |
|
|
Iron Absorption and Transport
|
Dietary iron (ferric Fe3+) is converted to (ferrous Fe2+) by stomach acid
Fe2+ is absorbed in duodenum and jejunum where it is oxidized back to Fe3+ Fe3+ combines with apoferritin to form ferritin OR Fe3+ combines with apotransferrin to form transferrin. Heme iron from meat is absorbed directly into the ileum. Hepcidin is a hormone secreted by liver, acts on small intestine and inhibits iron absorption. |
|
|
Chem tests for anemia
|
Serum iron : Total iron in serum
Serum ferritin : Acute phase reactant (increases during stress or inflammation) One of the most sensitive indicators of iron stores Transferrin : main way that iron is transported in blood Total iron binding capacity (TIBC) : Reflects the ability of transferrin to bind to iron If TIBC is high, then the amount of Fe currently bound is low. Transferrin saturation : (Serum iron/TIBC) 100 Reflects the amount of iron that is “bound” in the serum |
|
|
3 stages of iron deficiency
|
1. Depletion of iron stores
Decrease or absence of stainable iron (Prussian blue) in BM Decreased serum ferritin Increased TIBC 2. Iron-deficient erythropoiesis Decreased Hgb in developing RBC’s without frank anemia Early microcytosis Decreased transferrin saturation = (Serum iron/TIBC) 100 3. Iron-deficiency anemia Decreased hemoglobin synthesis with anemia and significant microcytosis and hypochromia Anisocytosis of the RBC’s Decreased serum iron, serum ferritin, transferrin saturation Increased TIBC |
|
|
Iron deficiency anemia (IDA)
|
Dec red count, hgb, hct, MCV, MCHC, serum iron, serum ferritin, transferrin sat.
Increased TIBC -funky nails, ragged erythoid precursors |
|
|
Anemia of inflammation
|
Red cell count, hemoglobin, and hematocrit all borderline low.
MCHC normal MCV slightly low Serum iron low Serum ferritin normal or increased TIBC decreased microcytic, normochromic |
|
|
Sideroblastic anemia
|
Accumulation of iron in the mitochondria
Some evidence for enzyme deficiencies in protporphyrin and heme sythesis Inherited -Sex-linked and autosomal recessive. -5-aminolevulinic synthetase deficient: Protoporphyrin ring cannot be formed. -Iron accumulates in the mitochondria, because it cannot be incorporated into heme. Acquired -Primary or idiopathic = myelodysplastic syndrome (RARS – refractory anemia with ringed sideroblasts.) -Secondary = alcohol, lead (impairs heme syth at several steps), TB drugs Dimorphic blood picture Pappenheimer bodies Siderocytes and ringed sideroblasts with Prussian blue (BM) Increased serum ferritin Increased serum iron TIBC is normal or low |
|
|
iron overload (hemochromatosis)
|
Iron overload Def: accumulation of excess iron in reticuloendothelial cells in various tissues.
Hemochromatosis Def: a clinical disorder that results in tissue damage resulting from excess iron. -Increase in intestinal iron absorption due to mutation in the HFE gene on chromosome 6 -Postulated: decreased hepcidin or hepcidin binding or mutated hepcidin receptor |
|
|
hereditary hematochromatosis
|
erythropoiesis is normal, so peripheral smear is usually normal;
Alanine and aspartate aminotransaminase (ALT, AST) are high, serum iron is high, serum ferritin is high, serum transferrin is high, TIBC usually normal, increased % transferrin saturation is a hallmark, presence of HFE gene mutation |
|
|
Thalassemia
|
Globin chain disorders
Microcytic process Hemolytic anemias Normal iron status Not linked to iron problems Can affect multiple organs May require transfusions Not enough functional hemoglobin A produced |
|
|
General Alpha vs beta thal
|
ALPHA
High incidence in Asian, Saudi, Filipino populations We inherit 2 alpha genes from each parent for a total of 4 alpha genes In alpha Thalassemia, there are 0, 1 or 2 functional genes. There is also a carrier state with 3 functional genes. Gene deletions are the reasons for lack of alpha genes BETA We inherit 1 beta gene from each parent for a total of 2 beta genes. In Beta Thalassemia we inherit defective beta genes Heterozygotes or homozygotes Beta 0 means no beta chain synthesis Beta + means there is a limited amount of beta chain synthesis 200 mutations to date Many populations affected Decreased number of beta chains = less Hb A |
|
|
Alpha thal
|
0- Barts hydrops fetalis (no α alpha chains) Hemoglobin Bart’s = (g4)
1- Hemoglobin H Dz. (1 functional alpha gene) Hemoglobin H formed instead of hemoglobin A Hgb H = (b4), unstable Hgb H inclusions (pitted golf ball Hgb = 10g/dL, retic = 5-10%, micro/hypo 2- Alpha thalassemia trait or Alpha Thal Minor (2 functional alpha chains) Mild anemia 3- Silent carrier (3 functional α chains) Hematologically normal |
|
|
osmotic fragility
|
Used to screen for carrier states of thalassemia (and also hereditary spherocytosis)
Add various dilutions of normal saline [.85% NaCl] to RBC’s -Norm RBC’s lyse @ 0.45% NaCl -HE spherocytes (decreased SA:volume) lyse at .65% NaCl. -Beta thal and alpha thal trait lyse at < 0.45% NaCl. -Heterozygous beta thal, Hb H, Alpha thal trait have decreased osmotic fragility. Recall that there are lots or target cells seen in thalassemia (increased cell SA:volume) |
|
|
Beta thalassemia major – AKA Cooley’s Anemia.
|
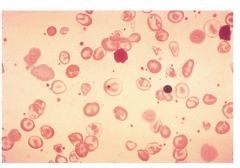
- Little or no beta chains (therefore little or no hemoglobin A)
-Point mutations disrupt gene expression for Beta chains -Infant is ok for first 6 months, b/c Hgb F is produced -Adults can try to compensate by increasing Hgb F production up to ***60-95% of all Hgb. -Hgb A2 = 2-7%. *** -Microcytic, hypochromic anemia -Polychromasia -NRBC -Changes in facial structures -Overexpansion of BM leads to thin bones/fractures -Low hemoglobin levels (6- 9 g/dl) --6 = transfusion lvl ; Transfusion (q 2-5 wks) may lead to increased iron overload and chelation may be necessary -Enlarged spleen is destroying defective RBCs |
|
|
Beta thalassemia minor/trait (1 abnormal beta gene)
|
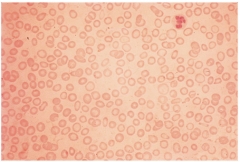
Point mutations disrupt gene expression for Beta chains
Mild microcytic, hypochromic anemia (HGB = 11-13g/dL) ****Hemoglobin A2 increased to 5-10% ****Hemoglobin F can increase to 2-5% Mimics IDA values but RBC count may be elevated due to bone marrow compensation Iron is NOT the problem in (Beta) Thal minor, so docs should not treat patients unnecessarily with iron! |
|
|
Hereditary Persistence of Fetal Hemoglobin (HPFH)
|
Sometimes due to increased Hb F related to decreased delta and beta chain synthesis (delta-beta thalassemia) and sometimes the gene for gamma chains just never got switched off.
Homozygous mimics thalassemia major. Heterozygous are hematologically normal. Seen in Africans and Greeks ***Use the Kleihauer-Betke acid elution test to screen for Hb F. |