Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
97 Cards in this Set
- Front
- Back
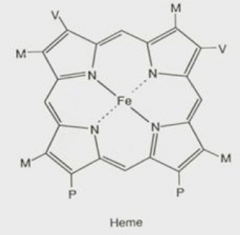
What are the functions of heme?
|
- Transport of O2 (on hemoglobin and myoglobin)
- Electron transport (respiratory cytochromes) - Oxidation-reduction reactions (cytochrome P450 enzymes) |
|
|
What are the sites of heme synthesis? What forms of heme are made in these locations?
|
- Bone Marrow - hemoglobin
- Liver - cytochrome P450 enzymes - Virtually all cells (except mature RBCs) - other important cellular proteins |
|
|
Where is hemoglobin synthesized? How much?
|
- Bone marrow
- 6-7g synthesized per day to replace heme lost through normal turnover of RBCs |
|
|
Where are cytochrome P450 enzymes synthesized? Function?
|
- Liver
- Drug detoxification |
|
|
Why can't hemoglobin be synthesized in mature erythrocytes?
|
They lack mitochondria and other organelles
|
|
|
What kind of molecule is Porphyrin? Use?
|
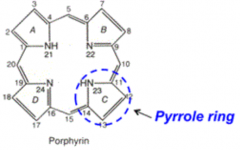
- Cyclic, predominantly planar tetrapyrroles
- Capable of chelating to various metals - Form prosthetic groups for various biological molecules |
|
|
What are the components of heme?
|
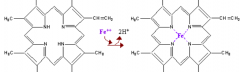
- Porphyrin derivative: Protoporphyrin IX
- Single ferrous iron (Fe2+ = reduced form) |
|
|
What porphyrin is found in heme? What is the name for this form when combined with ferrous (Fe2+) iron?
|
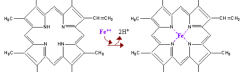
Protoporphyrin IX:
- Specific isomer of porphyrin that contains specific substituent groups on the four pyrrole rings - Substituent groups provide important sites for binding to its apoproteins Forms Ferroprotoporphyrin IX |
|
|
What happens if there is auto-oxidation of Ferroprotoporphyrin IX (heme)?
|
Forms Ferriprotoporphyrin IX (with an "i")
- Called "Hemin" - Contains ferric Fe3+ iron |
|
|
How many stages of heme biosynthesis are there? Where do these steps take place?
|
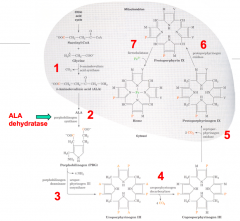
7 major steps:
- 1st and last 3 (5, 6, and 7) take place in mitochondria - 2, 3, and 4 take place in cytosol |
|
|
What are the molecules in the 7 steps to synthesize Heme?
|
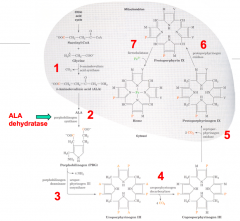
1. Succinyl CoA + Glycine
2. ALA: 5-Aminolevulinate 3a. PBG: Porphobilinogen 3b. Hydroxymethylbilane 4. Uroporphyrinogen III Isomer 5. Protoporphyrinogen IX 6. Protoporphyrin IX 7. Heme / Ferroprotoporphyrin IX |
|
|
What are the enzymes in the 7 steps to synthesize Heme?
|
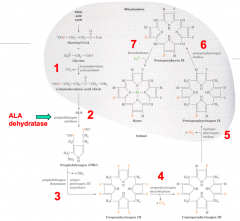
1. ALAS: 5-Aminolevulinate Synthase
2. ALAD: ALA Dehydratase 3a. PBGD: Porphobilinogen Deaminase 3b. UROS: Uroporphyrinogen III Cosynthase 4. UROD: Uroporphyrinogen Decarboxylase 5. CPO: Coproporphyrinogen III Oxidase 6. PPO: Protoporphyrinogen IX Oxidase 7. Ferrocheletase |
|
|
What happens in the first step of heme synthesis?
|
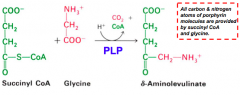
- Substrate: Succinyl-CoA + Glycine
- Enzyme: ALAS - 5-aminolevulinate synthase - Cofactors: PLP - Product: ALA - 5-aminolevulinate (+ CO2 + CoA) |
|
|
What happens in the second step of heme synthesis, after formation of ALA?
|
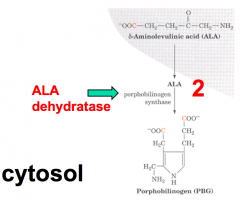
- Substrate: 2 x ALA - 5-aminolevulinate
- Enzyme: ALAD - 5-aminolevulinate dehydratase - Cofactors: Zn2+ - Product: PBG - Porphobilinogen |
|
|
What happens in part one of the third step of heme synthesis, after formation of PBG?
|
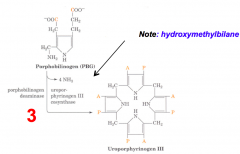
- Substrate: 4 x PBG - Porphobilinogen
- Enzyme: PBGD - Porphobilinogen Deaminase - Cofactors: - Product: Hydroxymethylbilane (+ NH3 x4) |
|
|
What happens in part two of the third step of heme synthesis, after formation of Hydroxymethylbilane?
|
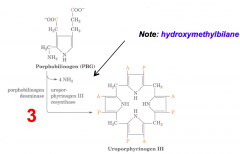
- Substrate: Hydroxymethylbilane
- Enzyme: UROS - Uroporphyrinogen III Cosynthase - Cofactors: - - Product: Uroporphyrinogen III Isomer |
|
|
What happens in the fourth step of heme synthesis, after formation of Uroporphyrinogen III Isomer?
|
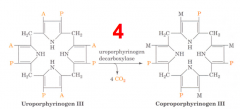
- Substrate: Uroporphyrinogen III Isomer
- Enzyme: UROD - Uroporphyrinogen Decarboxylase - Cofactors: - - Product: Coprophorphyrinogen III (+ CO2 x4) |
|
|
What happens in the fifth step of heme synthesis, after formation of Coprophorphyrinogen III?
|
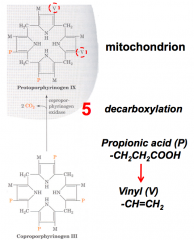
- Substrate: Coprophorphyrinogen III
- Enzyme: CPO - Coprophorphyrinogen III Oxidase - Cofactors: - - Product: Protoporphyrinogen IX |
|
|
What happens in the sixth step of heme synthesis, after formation of Protoporphyrinogen IX?
|
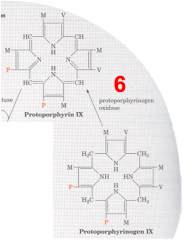
- Substrate: Protoporphyrinogen IX
- Enzyme: PPO - Protoporphyrinogen IX Oxidase - Cofactors: - - Product: Protoporphyrin IX |
|
|
What happens in the seventh step of heme synthesis, after formation of Protoporphyrin IX?
|
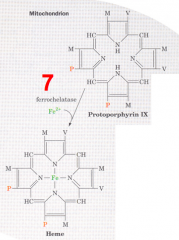
- Substrate: Protoporphyrin IX + Fe2+
- Enzyme: Ferrocheletase - Cofactors: - - Product: Heme |
|
|
Where do the carbon and nitrogen atoms of the porphyrin ring in heme originate?
|
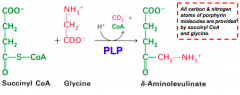
All C and N are from Succinyl-CoA and Glycine
|
|
|
What is the enzyme in the first step of heme synthesis? Location?
|
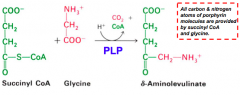
ALAS: 5-Aminolevulinate synthase
- Found in the inner mitochondrial membrane, but encoded by a nuclear gene family - Therefore, nascent protein must be imported into mitochondrion |
|
|
What does the enzyme 5-Aminolevulinate synthase (ALAS) require? What step? Mechanism?
|
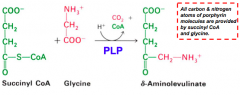
- Step 1
- Pyridoxal Phosphate (PLP) dependent enzyme - Condensation of glycine w/ succinyl-CoA takes place while amino group of glycine is in Schiff base linkage to PLP aldehyde; CoA and glycine carboxyl are lost during condensation |
|
|
What are the two forms of 5-Aminolevulinate synthase (ALAS)?
|
- ALAS1 - liver isoform
- ALAS2 - erythroid / reticulocyte isoform |
|
|
How is feedback of the two ALAS isoforms different?
|
- ALAS1 (liver) has feedback inhibitino by heme or hemin
- ALAS2 (RBCs) is not regulated by feedback repression |
|
|
How is ALAS1 regulated in the liver?
|
- Feedback inhibition by heme or hemin regulates heme biosynthesis in liver (ALAS1)
- Heme inhibits ALAS1 synthesis at both transcriptional and translational level and as its mitochondrial import - ~100 drugs or metabolites can stimulate ALAS1 - Many drugs are metabolized by cytochrome P450s in liver and thus increase synthesis of cytochrome P450 enzymes (increasing demand for heme) |
|
|
How is ALAS2 regulated in the RBCs?
|
- Heme biosynthesis is not regulated by feedback repression of ALAS2
- Heme stimulates synthesis of globin and ensures that heme and globin are synthesized in correct ratio for assembly of hemoglobin - Drugs that cause a marked elevation of ALAS1 (eg, phenobarbital) do not affect ALAS2 |
|
|
What is the first pathway intermediate to include a pyrrole ring? When is it synthesized?
|
- Porphobilinogen (PBG)
- Synthesized in step 2 in cytosol by ALA dehydratase (ALAD) |
|
|
What is required for ALA dehydratase (ALAD) activity in step 2 of heme synthesis?
|
Zn2+ complexed to an active site cysteine
|
|
|
What step of heme synthesis is affected by lead poisoning? How does it affect this step?
|
- Step 2: ALA Dehydratase (ALAD)
- ALAD requires a Zn2+ in the active site, lead and other heavy metals can displace Zn2+ and eliminate the catalytic activity |
|
|
What are the implications of lead poisoning?
|
- Increases ALA in urine (the substrate for step 2 of heme synthesis)
- Increases ALA in blood (causes neurological side effects) - Lead may also directly affect the nervous system - Clinical manifestations mimic acute porphyrias |
|
|
What can cause a buildup of ALA levels in the blood? What are the implications?
|
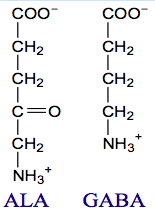
- Caused by lead poisoning (which eliminates catalytic activity of ALA dehydratase, ALAD)
- Toxic to brain perhaps because ALA has a similar structure as GABA; also ALA autoxidation generates reactive oxygen species (ROS) |
|
|
What happens structurally in the third step of heme synthesis?
|
Porphobilinogen Deaminase (PBGD)
- Head-to-tail condensation of four porphobilinogen molecules to form a linear tetrapyrrole (liberates 4 ammonium ions) Uroporphyrinogen III Cosynthase (UROS) - Directs the stereochemistry of the condensation reaction to yield Uroporphyrinogen III isomer |
|
|
What happens structurally in the fourth step of heme synthesis?
|
Decarboxylation of acetate side chains to methyl groups (by UROD)
|
|
|
What happens structurally in the fifth step of heme synthesis?
|
- Substrate (Coproporphyrinogen III) transported into intermembrane space
- Oxidase (CPO) converts specific propionic acid side chains to vinyl groups - Forms Protoporphyrinogen IX |
|
|
What happens structurally in the sixth step of heme synthesis?
|
Another mitochondrial oxidase (PPO) moves double bonds in the structure to form protoporphyrin IX
|
|
|
What happens structurally in the seventh step of heme synthesis?
|
Insertion of Fe2+ into Protoporphyrin IX to generate heme by Ferrochelatase
|
|
|
What inhibits Ferrocheletase (the last step of heme synthesis)?
|
- Lead poisoning
- Iron deficiency (anemia) - not enough Fe2+ to insert into Protoporphyrin IX |
|
|
What happens if there is an absence of Fe2+?
|
Ferrocheletase can insert Zn2+ into the protoporphyrin ring to yield a brilliantly fluorescent complex
|
|
|
What are porphyrias?
|
Defects in heme biosynthesis
|
|
|
What causes Porphyrias?
|
- Inherited / genetic disorders
- Acquired (rarely) disorders - Result from deficiency in specific enzymes of the porphyrin/heme biosynthetic pathway |
|
|
How do you classify porphyrias?
|
Based on the principal sites of heme biosynthesis and depending on the site of expression of the enzyme defect:
- Hepatic - Erythroid |
|
|
How are Porphyrias inherited?
|
* Autosomal dominant
- Exception: congenital erythropoietic porphyria which is autosomal recessive |
|
|
What is the most common porphyria?
|
Acute Intermittent Porphyria
|
|
|
What is the most common erythropoietic porphyria?
|
Erythropoietic Protoporphyria (EPP)
(also most common childhood porphyria) |
|
|
What is the most common porphyria in childhood?
|
Erythropoietic Protoporphyria (EPP)
(also most common erythropoietic porphyria) |
|
|
What type of pophyria is extremely rare?
|
Congenital Erythropoietic Porphyria (CEP)
|
|
|
What causes the clinical symptoms in porphyrias?
|
- Accumulation of intermediates upstream from the enzyme defect (measure in urine, blood, feces)
- Defects early in pathway (accumulation of ALA, prophobilinogen) result in neurologic dysfunction - Defects later in pathway (accumulation of cyclic tetrapyrroles, but not prophobilinogen) result in sunlight-induced cutaneous lesions; in presence of molecular O2, UV irradiation of cyclic tetrapyrroles generates ROS that can cause cellular damage |
|
|
What happens if there are defects early in the biosynthetic pathway for Heme?
|
Porphyria:
- Accumulation of ALA and prophobilinogen - Leads to neurological dysfunction |
|
|
What happens if there are defects later in the biosynthetic pathway for Heme?
|
Porphyria:
- Accumulation of cyclic tetrapyrroles - Sunlight-induced cutaneous lesions - In presence of molecular O2, UV irradiation of cyclic tetrapyrroles generates ROS that can produce cellular damage |
|
|
What are the two ways that Porphyrias can present?
|
Acute:
- Periodic acute attacks - Sx include abdominal pain, neurologic deficits, psychiatric symptoms, and reddish-colored urine Chronic: - Dermatologic diseases - May or may not include liver and nervous system |
|
|
What are some triggers that can bring about acute attacks of Porphyria?
|
- Nutritional changes (eg, hypoglycemia)
- Smoking - Certain drugs (barbiturates and sulfonamide antibiotics) - Steroid hormones, especially progesterone (some women develop attacks during second half of menstrual cycle when progesterone is high) |
|
|
What is true about Ferrochelatase?
a) found in cytosol b) rate-limiting enzyme in heme synthesis c) requires glycine for activity d) activity stimulated in presence of lead e) catalyzes last step of heme synthesis |
Catalyzes last step in heme synthesis
- Found in mitochondria - First step is rate-limiting - Does not require glycine for activity - Activity inhibited by lead and iron deficiency |
|
|
You are treating a patient with Porphyria Cutanea Tarda, most common porphyria. She came to your office w/ significant blistering on her hands following a day of gardening. Accumulation of what would cause photosensitivity?
|
Accumulation of cyclic tetrapyrroles:
4. Uroporphyrinogen III Isomer 5. Protoporphyrinogen IX 6. Protoporphyrin IX |
|
|
What is the function of Hemoglobin?
|
- Transport O2 from lungs (high O2 concentration) to peripheral tissues where O2 tension is low
- Transports some CO2 and H+ that are generated in peripheral tissues back to lungs (14% of CO2 made is carried on Hb) |
|
|
Why is it necessary for O2 to be carried on Hemoglobin? Consequences?
|
- O2 has a very low solubility in plasma (non-cellular part of blood)
- As a consequence, >98% of O2 that reaches the tissues is carried bound to hemoglobin in RBCs |
|
|
Why does CO2 not have to be carried on Hemoglobin?
|
- RBCs carry Carbonic Anhydrase which catalyzes the rapid reversible hydration of CO2 to H2CO3 which then dissociates to HCO3- and H+
- CO2 and HCO3- are soluble in plasma and RBC cytosol - Most of the CO2 made in tissues returns to lung as those species, but 14% bound to Hb |
|
|
What are the components of Hemoglobin?
|
Heterotetrameric protein: α2β2
- Contains 4 heme prosthetic groups responsible for binding O2 |
|
|
How does hemoglobin relate to myoglobin?
|
- Subunits of hemoglobin are evolutionarily related to myoglobin (monomeric protein abundant in muscle that is designed to store O2)
- Both proteins contain a heme prosthetic group (1/myoglobin and 4/hemoglobin) - Fe2+ is ferrous form of iron responsible for binding O2 - Hemoglobin has a sigmoidal (cooperative) binding curve, whereas Myoglobin has a hyperoblic binding curve to O2 |
|
|
How is hemoglobin affected by different forms of iron?
|
- Fe2+ (Ferrous form) - capable of binding O2
- Fe3+ (Ferric form) - cannot bind O2, inactive form called Methemoglobin (metHb) |
|
|
What kind of binding curve do Myoglobin and Hemoglobin have for O2? How do you explain the different binding curve for Hb?
|
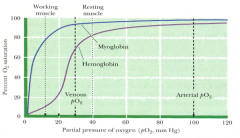
- Myoglobin: normal, hyperbolic binding curve
- Hemoglobin: sigmoidal, cooperative binding curve - D/t its more complex subunit structure, critical for its efficiency in loading O2 in lungs and unloading O2 in peripheral tissues |
|
|
What are the benefits of the cooperativity of O2 binding to hemoglobin?
|
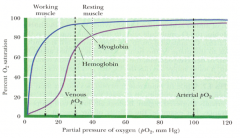
Allows Hb to release a much larger fraction of its own O2 load at the pO2 levels found in the blood of working and even resting muscle
|
|
|
How do the four subunits of hemoglobin lead to a cooperative/sigmoidal O2 binding relationship/
|
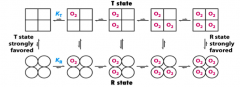
- Binding of O2 to one subunit induces a conformational change that is partially transmitted to adjacent subunits
- Transmission of partial conformational change induces an increased affinity for O2 by these adjacent subunits |
|
|
How does Carbon Monoxide compare to O2 binding to Hemoglobin?
|
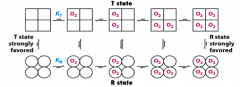
- CO has ~250-fold higher affinity for Hb than does O2
- When bound to the heme group of one subunit, it causes all four subunits to lock in the R conformation thereby limiting O2 release in peripheral tissues (needs to be in T state to release) |
|
|
What is the T state? What does it prefer? Same for R state?
|
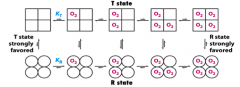
- T state = tense, favors dissociation (lower affinity for O2)
- R state = relaxed, favors association (higher affinity for O2) |
|
|
How does O2 binding change the conformation of a Hb subunit?
|
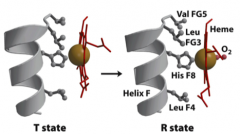
- Without O2 bound, the heme Fe2+ is pulled away from the plane of the porphyrin ring by a His residue of the Hb polypeptide chain (a His ring N is bound to the Fe2+)
- When O2 binds, it pulls the Fe2+ back into the plane of the ring and that moves the His residue and its whole section of the polypeptide chain - That in turn causes the Hb subunits to shift relative to one another to an arrangement that favors the R-conformation |
|
|
What is an allosteric regulator?
|
Molecule that can bind to a protein and induce a conformational change that alters the affinity for substrate (or ligand such as O2) at some other site (allo means other)
|
|
|
What are the allosteric regulators of O2 binding to Hb? Effects?
|
All bind to Hb and reduce its affinity for O2:
- Protons (H+) - CO2 - 2,3-Diphosphoglycerate (DPG) |
|
|
What are the effects of high amounts of H+, CO2, or 2,3-DPG?
|
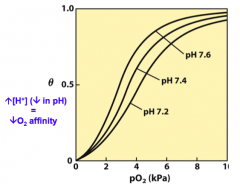
When these are present, the curve shifts to the right and O2 comes off of Hb
|
|
|
What are the normal values for:
- pH? - pO2? - pCO2? - HCO3-? |
- pH: 7.35-7.45
- pO2: 80-100 mmHg - pCO2: 35-45 mmHg - HCO3-: 22-26 mM |
|
|
What kind of effectors are H+ and CO2 on Hb binding of O2?
|
Heterotropic Negative Allosteric Effectors that decrease the affinity of Hb for O2
- Heterotropic: they are not O2 - Negative: decrease affinity for O2 - Allosteric: bind to a site other than O2 site |
|
|
What kind of effector is O2 on Hb binding of O2?
|
Homotropic Positive Allosteric Effector
- Homotropic: it is O2 - Positive: increases affinity for more O2 - Allosteric: binds to a site other than the site the next O2 could bind |
|
|
What kind of effector is O2 on Hb binding of H+ and CO2?
|
Heterotropic Negative Allosteric Effector
- Heterotropic: not CO2 or H+ - Negative: decreases affinity for CO2 and H+ - Allosteric: binds to a site different than they would bind |
|
|
What is the term for the reciprocal relationship between O2 and H+ binding to hemoglobin?
|
Bohr effect or isohydric shift
- Changes in H+ binding result from a shift in the pKa of specific residues (mostly histidines) d/t microenvironment effects triggered by conformational changes in Hb structure |
|
|
What is the mechanism of how increased H+ (↓pH) affects O2 binding?
|
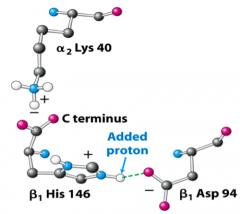
- In deoxyHb negative charge on Asp94 is near His146
- It is energetically favorable for N to be protonated (its pKa is higher) - Having His protonated in turn makes it favorable for Asp to stay near it, increasing the stability of the deoxyHb (T-state) - Conformation changes in going to oxyHb (R state) move His146 and Asp94 apart, pKa drops and H+ comes off |
|
|
What kind of effector is DPG on Hb binding of O2? Mechanism?
|
Heterotropic Negative Allosteric Effector
- Binds to a specific site in central cavity between β subunits by ionic interactions - This binding stabilizes the T state of deoxyHb |
|
|
What effects does DPG have on the O2 binding curves?
|
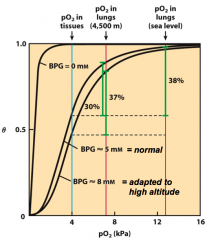
1. Without any DBG, Hb would be more like myoglobin (hyperbolic) and nearly useless for delivering O2 from lungs to tissues
2. DPG levels increase at high altitudes - There is less O2 at high altitudes, so tissues tend to become somewhat hypoxic - Increasing DPG lets RBC adapt to hypoxia by making it easier for O2 to dissociate from Hb |
|
|
What situations/conditions stimulate DPG release?
|
Causes of tissue hypoxia:
- High altitude - Smoking - Anemia |
|
|
How long does it take to make changes in DPG? Implications?
|
- A few days
- Therefore takes a few days to adapt to high altitude - Until then, strenuous aerobic exercise is difficult (more dyspnea) |
|
|
How does temperature affect O2 association/dissociation?
|
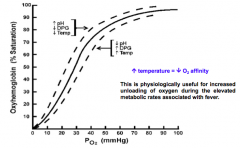
↑T → ↓O2 affinity
- During fever there are elevated metabolic rates so need increasing unloading of O2 |
|
|
What are the developmental forms of hemoglobin?
|
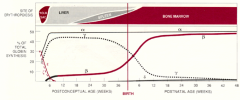
- ~2% HbF - Fetal: α2γ2 (until ~5-6 months)
- ~95% HbA1 - Adult 1: α2β2 (majority) - ~3% HbA2 - Adult 2: α2δ2 (very low) |
|
|
Which chromosome contains the α-globin gene(s)?
|
Chromosome 16 has two α-globin genes (total of 4 / person)
|
|
|
Which chromosome contains the β-globin gene(s)?
|
Chromosome 11 has a single β-globin gene (total of 2 / person)
|
|
|
Where is hemoglobin synthesized during the lifetime?
|
- Yolk Sac
- Liver - Spleen - Bone marrow |
|
|
How do the different forms of hemoglobin (α2β2, α2δ2, and α2γ2) differ?
|
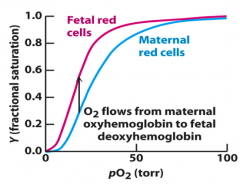
Binding affinity for O2
- Higher affinity of fetal Hb means fetus' circulation can draw O2 from maternal blood at pO2 present in placenta |
|
|
How is Sickle Cell Anemia inherited? Cause?
|
Homozygous recessive disease
- Point mutation in adult β-globin gene that causes substitution of valine for glutamic acid at amino acid 6 - Patients mainly contain HbS |
|
|
What is the most common hemoglobinopathy?
|
Sickle Cell Anemia
|
|
|
What are the effects of the mutation in Sickle Cell Anemia?
|
- Valine substituted for glutamic acid at position 6 is on the surface of the β-chain and it should be hydrophilic
- Valine is hydrophobic and its presence creates a sticky patch on deoxyHb that leads to polymerization of Hb tetramers into long chains - Val6 makes critical contact with hydrophobic acceptor pocket of β-subunit of another molecule formed by Leu88 and Phe85 - Intracellular fibers cause sickle cell shape and reduced deformability of RBCs that leads to problems w/ their passage through microcirculation |
|
|
What determines the amount/rate of polymerization of HbS?
|
- Degree of deoxygenation (can be affected by pH, ionic strength, and temperature); deoxyHbS forms insoluble polymers
- Intracellular Hb concentration - Relative amount of HbF present (HbF inhibits polymerization d/t Glu87 on γ-chain |
|
|
What occurs in a Thalassemia?
|
Inherited mutations cause a decreased synthesis of adult hemoglobin (α2β2)
|
|
|
What kind of mutations cause β-thalassemias?
|
- β° mutations have absent β-globin chain synthesis
- β⁺ mutations have reduced (but detectable) β-globin chain synthesis |
|
|
What are the implications of β-thalassemias?
|
- Deficit in HbA
- Unpaired α-chains precipitate in RBC precursors, resulting in apoptosis |
|
|
What are the types of β-thalassemias? Differences?
|
- β-thalassemia major: two β-thalassemia alleles w/ severe, transfusion-dependent anemia
- β-thalassemia minor: heterozygotes have only one β-thalassemia allele and mild or asymptomatic microcytic anemias |
|
|
What causes α-thalassemias?
|
Mutations that result in reduced or absent synthesis of α-globin chains
|
|
|
What are the implications of α-thalassemias?
|
Unpaired β-chains are more soluble than unpaired α-chains, thus effects less severe than in β-thalassemias
|
|
|
What is true of Oxygen:
a) binding converts Hb to T state b) dissociation from Hb is inhibited in patients w/ fever c) dissociation from Hb is enhanced at pH values below pH 7.4 d) binding to Hb causes conversion of a β strand to a random coil e) dissociation from Hb is inhibited in presence of CO2 |
Dissociation from Hb is enhanced at pH values below 7.4
|
|
|
What is true about Methemoglobin?
a) similar O2 association/dissociation curve as myoglobin b) similar O2 association/dissociation curve as hemoglobin c) monomer d) cannot bind hemoglobin e) contains ferric iron |
Cannot bind hemoglobin (it contains ferric / Fe3+ form)
|