![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
253 Cards in this Set
- Front
- Back
|
Principal sistema responsável pela produção sanguínea na vida fetal: |
Sistema retículo endotelial. |
|
|
Na vida adulta, caso haja uma doença muito grave, * pode ser acionado para a produção sanguínea. |
Sistema retículo endotelial. |
|
|
Para a eritropoiese são necessários principalmente 4 itens: |
Eritropoietina, ferro, ác fólico e vB12. |
|
|
Precursor da hemácia liberado pela MO: Aonde ocorre sua maturação? Quanto tempo demora? |
Reticulócito. Sistema circulatório. 1 a 2 dias. |
|
|
Destruição fisiológica da hemácia pelo baço: |
Hemocaterese. |
|
|
Destruição da hemácia de forma prematura: |
Hemólise. |
|
|
Primeiro item a ser investigado diante de uma anemia: A partir disso, há divisão em: |
Reticulócitos. Hipo/ hiperproliferativa. |
|
|
Tipo de anemia na qual há > 2% de retículócitos: E se <= 2%? |
Hiperproliferativa. Hipoproliferativa. |
|
|
A anemia hiperproliferativa só é encontrada em 2 situações: |
Anemia hemolítica e sg agudo. |
|
|
Segundo item a ser avaliado diante de uma anemia: |
Morfologia (VCM e HCM). |
|
|
Segundo item a ser avaliado diante de uma anemia: |
Morfologia (VCM e HCM). |
|
|
Tipo de anemia mais frequente: |
Ferropriva. |
|
|
Segundo item a ser avaliado diante de uma anemia: |
Morfologia (VCM e HCM). |
|
|
Tipo de anemia mais frequente: |
Ferropriva. |
|
|
A carencia nutricional (seja Fe, B12 ou ác fólico) leva a sintomas como: |
Glossite e queilite angular. |
|
|
Segundo item a ser avaliado diante de uma anemia: |
Morfologia (VCM e HCM). |
|
|
Tipo de anemia mais frequente: |
Ferropriva. |
|
|
A carencia nutricional (seja Fe, B12 ou ác fólico) leva a sintomas como: |
Glossite e queilite angular. |
|
|
Segundo item a ser avaliado diante de uma anemia: |
Morfologia (VCM e HCM). |
|
|
Tipo de anemia mais frequente: |
Ferropriva. |
|
|
A carencia nutricional (seja Fe, B12 ou ác fólico) leva a sintomas como: |
Glossite e queilite angular. |
|
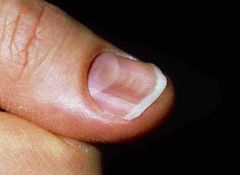
Front (Term) |
Coiloníquia. |
|
|
Sintomas causados especificamente pela falta de ferro: |
Pica, coiloníquia e disfagia. |
|
|
Local de absorção do ferro no corpo: |
I. delgado proximal. |
|
|
Proteína acoplada ao ferro no seu transporte: Onde é produzida? |
Transferrina. Fígado. |
|
|
Proteína acoplada ao ferro no seu transporte: Onde é produzida? |
Transferrina. Fígado. |
|
|
Paciente com 40/50 anos que passa a apresentar anemia ferropriva deve ser investigado para: |
Neoplasia GI. |
|
|
Proteína acoplada ao ferro no seu transporte: Onde é produzida? |
Transferrina. Fígado. |
|
|
Paciente com 40/50 anos que passa a apresentar anemia ferropriva deve ser investigado para: |
Neoplasia GI. |
|
|
Primeiro item do hemograma a apresentar alteração em uma anemia ferropriva: |
Ferritina (queda abaixo de 30). |
|
|
Plaquetas em fase avançada da anemia ferropriva: |
Elevadas (trombocitose). |
|
|
Plaquetas em fase avançada da anemia ferropriva: |
Elevadas (trombocitose). |
|
|
Principais causas de anemia ferropriva em crianças: E adultos: |
Prematuridade, desmame, ancilostomíase. Gravidez, hipermenorreia, má-absorção (dç celíaca), perda crônica de sangue pelo TGI. |
|
|
Dose de ferro elementar para reposição em anemia ferropriva (adulto e criança): |
A: 120-200mg/dia. C: 3-5mg/kg/dia. |
|
|
A resposta inicial ao tratamento da anemia ferropriva aparece com quantos dias? O que é observado? |
3/4 dias. Aumento de reticulócitos. Obs: pico do aumento ocorre entre 7 a 10d. |
|
|
Média de tempo para normalização após tratamento para anemia ferropriva: Por quanto tempo deve ser mantido o tto após normalização? |
2m. Mais 6m ou quando ferritina >15 (criança) ou >50 (adulto). |
|
|
Média de tempo para normalização após tratamento para anemia ferropriva: Por quanto tempo deve ser mantido o tto após normalização? |
2m. Mais 6m ou quando ferritina >15 (criança) ou >50 (adulto). |
|
|
Ferritina e TIBC na anemia ferropriva: |
Baixa e alta. |
|
|
Média de tempo para normalização após tratamento para anemia ferropriva: Por quanto tempo deve ser mantido o tto após normalização? |
2m. Mais 6m ou quando ferritina >15 (criança) ou >50 (adulto). |
|
|
Ferritina e TIBC na anemia ferropriva: |
Baixa e alta. |
|
|
Ferritina e TIBC na anemia de dç crônica: |
Alta e baixa. |
|
|
VCM e HCM mais comum na anemia de dç crônica: |
Normo/normo. |
|
|
VCM e HCM mais comum na anemia de dç crônica: |
Normo/normo. |
|
|
Tratamento da anemia de dç crônica: |
Tratar doença de base. |
|
|
Substância que se acumula quando não há ativação normal do ácido fólico pela B12: |
Homocisteína. |
|
|
Neutrófilos na anemia megaloblástica: |
Hipersegmentados: 5 ou mais núcleos (patognomônico). |
|
|
LDH e BI na anemia megaloblástica: |
Aumentados. |
|
|
LDH e BI na anemia megaloblástica: |
Aumentados. |
|
|
Local de absorção do ác fólico: |
Intestino delgado proximal. |
|
|
LDH e BI na anemia megaloblástica: |
Aumentados. |
|
|
Local de absorção do ác fólico: |
Intestino delgado proximal. |
|
|
Causas da deficiência de folato (4): |
Má nutrição: alcoolismo. Aumento da necessidade: gestante, hemólise crônica. Diminuição de absorção: dç celíaca, fenitoína. Diminuição da regeneração (reativação do ác fólico após ser usado): MTX. |
|
|
Local onde há separação da vB12/ligante R e ligação do fator intrínseco à vB12: |
Duodeno. |
|
|
Local de absorção de vB12/ fator intrínseco: |
Íleo distal. |
|
|
Quadro clínico da anemia megaloblástica: |
Sd anêmica + glossite, queilite e diarreia (perda de renovação celular do TGI). |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
Causa da anemia sideroblástica: |
Deficiência de protoporfirina (hereditária ou adquirida - álcool, diminuição de B6, chumbo). |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
Causa da anemia sideroblástica: |
Deficiência de protoporfirina (hereditária ou adquirida - álcool, diminuição de B6, chumbo). |
|
|
Laboratório da anemia sideroblástica: |
Anemia micro/micro, aumento de ferro, ferritina e sat de transferrina. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
Causa da anemia sideroblástica: |
Deficiência de protoporfirina (hereditária ou adquirida - álcool, diminuição de B6, chumbo). |
|
|
Laboratório da anemia sideroblástica: |
Anemia micro/micro, aumento de ferro, ferritina e sat de transferrina. |
|
|
A anemia sideroblástica está relacionada a risco de: |
Hemocromatose. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
Causa da anemia sideroblástica: |
Deficiência de protoporfirina (hereditária ou adquirida - álcool, diminuição de B6, chumbo). |
|
|
Laboratório da anemia sideroblástica: |
Anemia micro/micro, aumento de ferro, ferritina e sat de transferrina. |
|
|
A anemia sideroblástica está relacionada a risco de: |
Hemocromatose. |
|
|
Mielograma de indivíduo com anemia sideroblástica demonstra: |
Sideroblasto em anel. |
|
|
A deficiência de * pode gerar manifestações neurológicas na anemia megaloblástica. |
VB12. |
|
|
A diminuição de vB12 leva ao acúmulo de * porque ele deixa de ser convertido em **. |
* ác metilmalônico. ** succinil-coa. |
|
|
Laboratório na anemia megaloblástica: |
Anemia macrocítica + aumento de neutrófilos. Diminuição das plaquetas e leucócitos. Aumento de LDH e BI. |
|
|
Tratamento da anemia megaloblástica: |
Def de B12: reposição IM. Def de AF: 1 - 5mg/dia VO. Atenção para hipocalemia (acompanhar na 1a semana) e na reposição de AF -> pode mascarar def de B12 (melhora anemia mas não o quadro neurológico). |
|
|
Causa da anemia sideroblástica: |
Deficiência de protoporfirina (hereditária ou adquirida - álcool, diminuição de B6, chumbo). |
|
|
Laboratório da anemia sideroblástica: |
Anemia micro/micro, aumento de ferro, ferritina e sat de transferrina. |
|
|
A anemia sideroblástica está relacionada a risco de: |
Hemocromatose. |
|
|
Mielograma de indivíduo com anemia sideroblástica demonstra: |
Sideroblasto em anel. |
|
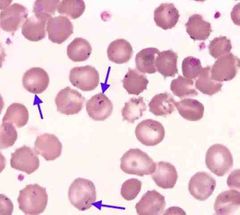
Indivíduo com anemia sideroblástica, qual é o achado? |
Corpúsculos de Pappenheimer. |
|
|
Tratamento de anemia sideroblástica: |
Causa adquirida: retirar causa. Hereditário: vB6 (resultados +-). |
|
|
Hemocaterese: |
Morte fisiológica das hemácias (120d). |
|
|
Diante de hemólise, o corpo pode fazer sua compensação através de: |
hiperplasia da medula, estoques de ferro, b12 e folato. |
|
|
Diante de hemólise, o corpo pode fazer sua compensação através de: |
hiperplasia da medula, estoques de ferro, b12 e folato. |
|
|
Causas de reticulocitose: |
Anemia hemolítica, sangramento agudo, melhora da anemia ferropriva. |
|
|
Diante de hemólise, o corpo pode fazer sua compensação através de: |
hiperplasia da medula, estoques de ferro, b12 e folato. |
|
|
Causas de reticulocitose: |
Anemia hemolítica, sangramento agudo, melhora da anemia ferropriva. |
|
|
Clínica de uma anemia hemolítica: |
Síndrome anêmica, esplenomegalia, icterícia leve, cálculo biliar. |
|
|
Diante de hemólise, o corpo pode fazer sua compensação através de: |
hiperplasia da medula, estoques de ferro, b12 e folato. |
|
|
Causas de reticulocitose: |
Anemia hemolítica, sangramento agudo, melhora da anemia ferropriva. |
|
|
Clínica de uma anemia hemolítica: |
Síndrome anêmica, esplenomegalia, icterícia leve, cálculo biliar. |
|
|
Lab na anemia hemolítica: |
Anemia normo, aumento de VCM, aumento de LDH, aumento de bilirrubina indireta e diminuição da haptoglobina. |
|
|
Anemião + reticulocitopenia: |
Crise aplásica e megaloblástica. |
|
|
Vírus que infecta o eritroblasto, causando reticulocitopenia em pacientes com hemólise: |
Parvovírus B19 (eritema infeccioso). |
|
|
Vírus que infecta o eritroblasto, causando reticulocitopenia em pacientes com hemólise: |
Parvovírus B19 (eritema infeccioso). |
|
|
Tratamento da crise aplásica: |
Suporte + avaliar necessidade de transfusão. |
|
|
Vírus que infecta o eritroblasto, causando reticulocitopenia em pacientes com hemólise: |
Parvovírus B19 (eritema infeccioso). |
|
|
Tratamento da crise aplásica: |
Suporte + avaliar necessidade de transfusão. |
|
|
Motivo da reticulocitopenia na crise megaloblástica: Tratamento: |
Perda de matéria prima: ác fólico. Prevenção: todo paciente com hemólise deve tomar ác fólico diariamente. |
|
|
Vírus que infecta o eritroblasto, causando reticulocitopenia em pacientes com hemólise: |
Parvovírus B19 (eritema infeccioso). |
|
|
Tratamento da crise aplásica: |
Suporte + avaliar necessidade de transfusão. |
|
|
Motivo da reticulocitopenia na crise megaloblástica: Tratamento: |
Perda de matéria prima: ác fólico. Prevenção: todo paciente com hemólise deve tomar ác fólico diariamente. |
|
|
Como diferenciar anemia megaloblástica e crise megaloblástica? |
Anemia megaloblástica tem neutrófilos hipersegmentados. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
A presença de Coombs direto + e microesferócitos indica: |
Anemia hemolítica autoimune. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
A presença de Coombs direto + e microesferócitos indica: |
Anemia hemolítica autoimune. |
|
|
Dois principais grupos de anemia hemolítica autoimune: |
Anticorpos quentes (IgG): 75%. Frios (IgM). |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
A presença de Coombs direto + e microesferócitos indica: |
Anemia hemolítica autoimune. |
|
|
Dois principais grupos de anemia hemolítica autoimune: |
Anticorpos quentes (IgG): 75%. Frios (IgM). |
|
|
Principais drogas relacionadas com a anemia hemolítica autoimune: |
Penicilina e metildopa. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
A presença de Coombs direto + e microesferócitos indica: |
Anemia hemolítica autoimune. |
|
|
Dois principais grupos de anemia hemolítica autoimune: |
Anticorpos quentes (IgG): 75%. Frios (IgM). |
|
|
Principais drogas relacionadas com a anemia hemolítica autoimune: |
Penicilina e metildopa. |
|
|
Tratamento da anemia hemolítica autoimune por anticorpos quentes: |
Tratar causa, Corticoide/ rituximab, esplenectomia. |
|
|
Anemião + injúria renal aguda: |
Crise hiperemolítica. |
|
|
Doenças que causam anemia hemolítica autoimune por IgM: Tratamento: |
Micoplasma, mieloma, macroglobulinemia. Tratar a causa, avaliar rituximab. |
|
|
Causas da crise hiperemolítica: |
Diminuição da G6PD e picada da aranha marrom (Loxocelles). |
|
|
O que será encontrado no paciente com crise hiperemolítica? |
Hemoglobinúria maciça com lesão renal -> NTA com IRA oligúrica. |
|
|
Anemião + bação: Qual sua principal causa? |
Sequestro esplênico. Anemia falciforme. |
|
|
Anemias hemolíticas hereditárias: |
Esferocitose, G6PD, hemoglobinopatias (falciforme, talassemia). |
|
|
Anemias hemolíticas adquiridas: |
Hemoglobinúria paroxística noturna, hiperesplenismo, SHU/PTT. |
|
|
A presença de Coombs direto + e microesferócitos indica: |
Anemia hemolítica autoimune. |
|
|
Dois principais grupos de anemia hemolítica autoimune: |
Anticorpos quentes (IgG): 75%. Frios (IgM). |
|
|
Principais drogas relacionadas com a anemia hemolítica autoimune: |
Penicilina e metildopa. |
|
|
Tratamento da anemia hemolítica autoimune por anticorpos quentes: |
Tratar causa, Corticoide/ rituximab, esplenectomia. |
|
|
Clínica da esferocitose: |
Criança + esplenomegalia + aumento de CHCM + esferócitos. |
|
|
Diagnóstico e tratamento da esferocitose: |
Teste da fragilidade osmótica. Esplenectomia após os 5 anos SE anemia grave (Hb<8; ret>10%) e/ou repercussões clínicas (não cresce, não realiza algumas atividades). |
|
|
Em um paciente portador de esferocitose esplenectomizado continuará a haver esferócitos: V/F. |
V. |
|
|
Paciente portador de esferocitose que foi esplenectomizado continuará a fazer hemólise, porém com sintomas mais brandos: V/F. |
F. |
|
|
Deficiência de G6PD é mais comum em qual sexo? Quais são as causas dessa doença? |
Masculino. Infecções (causa mais comum), drogas (sulfa, primaquina, dapsona, naftalina e nitrofurantoína). |
|
|
Anemia hemolítica que acontece após superoxidação: |
Deficiência de G6PD. |
|
|
Anemia hemolítica que acontece após superoxidação: |
Deficiência de G6PD. |
|
|
Diagnóstico e tratamento da deficiência de G6PD: |
Corpúsculo de Heinz ou medida da atividade de G6PD. Tratamento: apenas prevenção - não tomar as drogas, procurar atendimento se febre. |
|
|
A hemoglobinúria paroxística noturna é uma desordem genética que ocorre na 2a semana de vida: V/F. |
F: desordem genética adquirida extraútero. |
|
|
A hemoglobinúria paroxística noturna é uma desordem genética que ocorre na 2a semana de vida: V/F. |
F: desordem genética adquirida extraútero. |
|
|
Doença que tem como fisiopatologia uma mutação que deixa a hemácia mais fácil de ser lisada pelo complemento: |
Hemoglobinúria paroxística noturna. |
|
|
A hemoglobinúria paroxística noturna é uma desordem genética que ocorre na 2a semana de vida: V/F. |
F: desordem genética adquirida extraútero. |
|
|
Doença que tem como fisiopatologia uma mutação que deixa a hemácia mais fácil de ser lisada pelo complemento: |
Hemoglobinúria paroxística noturna. |
|
|
Tríade clássica da hemoglobinúria paroxística noturna: |
Trombose abdominal, pancitopenia e hemólise. |
|
|
A hemoglobinúria paroxística noturna é uma desordem genética que ocorre na 2a semana de vida: V/F. |
F: desordem genética adquirida extraútero. |
|
|
Doença que tem como fisiopatologia uma mutação que deixa a hemácia mais fácil de ser lisada pelo complemento: |
Hemoglobinúria paroxística noturna. |
|
|
Tríade clássica da hemoglobinúria paroxística noturna: |
Trombose abdominal, pancitopenia e hemólise. |
|
|
Diagnóstico e tratamento da hemoglobinúria paroxística noturna: |
Citometria de fluxo (diminuição de CD55/ CD59). Tratamento: eculizumab (inibe o complemento - muito caro). |
|
|
Ap receber oxigênio, o drepanócito jovem volta a ter formato normal: V/F. |
V. |
|
|
Ap receber oxigênio, o drepanócito jovem volta a ter formato normal: V/F. |
V. |
|
|
idade do início da Clínica da anemia falciforme: |
Início aos 6m (HbF alta até então) |
|
|
Ap receber oxigênio, o drepanócito jovem volta a ter formato normal: V/F. |
V. |
|
|
idade do início da Clínica da anemia falciforme: |
Início aos 6m (HbF alta até então) |
|
|
A crise vaso oclusiva da anemia falciforme pode se manifestar por: |
1) sd mão pé; 2) crise óssea; 3) sequestro esplênico; 4) sd torácica aguda; 5) AVE. |
|
|
Dois principais sistemas nos quais haverá lesão crônica na anemia hemolítica: |
Renal e ósseo. |
|
|
Dois principais sistemas nos quais haverá lesão crônica na anemia hemolítica: |
Renal e ósseo. |
|
|
Normalmente a primeira manifestação da anemia falciforme é: Com que idade ocorre? Qual sua clínica? |
Sd mão pé. Até 2 a 3 anos. Dor com dactilite. |
|
|
Dois principais sistemas nos quais haverá lesão crônica na anemia hemolítica: |
Renal e ósseo. |
|
|
Normalmente a primeira manifestação da anemia falciforme é: Com que idade ocorre? Qual sua clínica? |
Sd mão pé. Até 2 a 3 anos. Dor com dactilite. |
|
|
Raio x na síndrome mão pé: |
Normal (rx não vê isquemia óssea). |
|
|
Crise álgica/ osteoarticular mais comum na anemia falciforme: Ocorre em que idade? Clínica? |
Crise óssea. >3 anos. Dor sem dactilite. Doi mais aonde há mais medula (ossos longos). |
|
|
Crise álgica/ osteoarticular mais comum na anemia falciforme: Ocorre em que idade? Clínica? |
Crise óssea. >3 anos. Dor sem dactilite. Doi mais aonde há mais medula (ossos longos). |
|
|
Porque o sequestro esplênico normalmente só ocorre até os 5 anos em paciente com anemia falciforme? |
Muita fibrose (autoesplenomegalia). |
|
|
Principal causa de morte em paciente com anemia falciforme até os 5 anos: |
Sepse por germe encapsulado. |
|
|
Deve ser feita esplenectomia cirúrgica em paciente com anemia falciforme após 2o episódio de sequestro esplênico: V/F. |
F: após a primeira crise. |
|
|
Em pacientes com anemia falciforme, o AVE hemorrágico é mais comum em crianças e o isquêmico, em adultos: V/F. |
F: contrário. |
|
|
Lesão renal crônica na anemia falciforme: |
Necrose de papila, GEFS, câncer da medula renal. |
|
|
Lesão renal crônica na anemia falciforme: |
Necrose de papila, GEFS, câncer da medula renal. |
|
|
Lesão óssea crônica na anemia falciforme: |
Osteopenia, necrose de cabeça do fêmur, osteomielite por Salmonella/ S aureus. |
|
|
Lesão renal crônica na anemia falciforme: |
Necrose de papila, GEFS, câncer da medula renal. |
|
|
Lesão óssea crônica na anemia falciforme: |
Osteopenia, necrose de cabeça do fêmur, osteomielite por Salmonella/ S aureus. |
|
|
Tratamento da crise vaso oclusiva na anemia falciforme: |
Hidratação (não fazer excessiva). Oxigênio se sat <92%. Analgesia incluindo opioides. ATB: B lactâmico (+macrolídeo) para pacientes com febre >=38,5 hipotensão arterial, Hb <5 e leuco >30000. |
|
|
Tratamento crônico na anemia falciforme: |
Ác fólico, vacinação, penicilina VO 2-3m até os 5a, internar se febre. Hidroxiureia se sd álgica, prevenção secundária à sd torácica aguda e AVE ou anemia grave sintomática. Tx MO: curativo. Ideal que seja feito em <16a. |
|
|
Mecanismo de atuação da hidroxiureia: |
Aumenta HbF. |
|
|
Quando transfundir paciente com anemia falciforme? |
Criança: anemia sintomática/ grave -> Hb <5,5. Adultos: Hb <6. |
|
|
Quando transfundir paciente com anemia falciforme? |
Criança: anemia sintomática/ grave -> Hb <5,5. Adultos: Hb <6. |
|
|
Quando fazer exsanguíneo transfusão aguda em paciente com anemia falciforme? |
Quadros graves (AVE, sd torácica aguda). |
|
|
Quando transfundir paciente com anemia falciforme? |
Criança: anemia sintomática/ grave -> Hb <5,5. Adultos: Hb <6. |
|
|
Quando fazer exsanguíneo transfusão aguda em paciente com anemia falciforme? |
Quadros graves (AVE, sd torácica aguda). |
|
|
Quando fazer exsanguíneo transfusão regular em paciente com anemia falciforme? |
Caso não haja resposta a hidroxiureia e prevenção secundária a AVE e sd torácica aguda. Alvo: hb 9-10 e hbs <30%. |
|
|
Na talassemia major não há HbA: V/F. |
V. |
|
|
Clínica e tratamento da talassemia major: |
Anemia grave, alterações ósseas, hepatoesplenomegalia, hemocromatose. Transfusão crônica, quelante de ferro, esplenectomia e tx MO. |
|
|
Clínica e tratamento da talassemia major: |
Anemia grave, alterações ósseas, hepatoesplenomegalia, hemocromatose. Transfusão crônica, quelante de ferro, esplenectomia e tx MO. |
|
|
Principal causa de morte na talassemia major: |
Hemocromatose. |
|
|
Não há hemólise na talassemia minor: v/f. |
V. |
|
|
Clínica e tratamento da talassemia minor: |
Assintomática. Se anemia, hipo/micro. Aconselhamento genético. |
|
|
Clínica e tratamento da talassemia minor: |
Assintomática. Se anemia, hipo/micro. Aconselhamento genético. |
|
|
Lab da talassemia minor: |
Anemia leve porém muito micro (VCM < 75). RDW normal. Cinética de ferro normal. |
|
|
Clínica e tratamento da talassemia minor: |
Assintomática. Se anemia, hipo/micro. Aconselhamento genético. |
|
|
Lab da talassemia minor: |
Anemia leve porém muito micro (VCM < 75). RDW normal. Cinética de ferro normal. |
|
|
RDW na talassemia minor e na anemia ferropriva: |
Normal. Aumentado. |
|
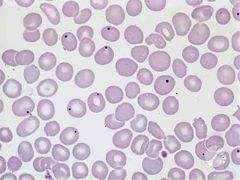
O que estamos vendo? Em que doença ocorrem? |
Corpúsculos de Howell-Jolly. Talassemia major. |
|
|
Paciente com traço falcêmico só faz crise álgica diante de infecção grave: V/F. |
F: assintomático do ponto de vista hematológico. |
|
|
Exame padrão ouro para o diagnóstico de ferropenia: |
Aspirado de medula óssea com corante azul da prússia. Se não houver ferro corável estamos diante de um diagnóstico 100% acurado de ferropenia. |