![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
191 Cards in this Set
- Front
- Back
|
Codominance |
Both alleles contributes to the phenotype of the heterzygotes Eg blood group HLA groups |
|
|
Variable expressivity |
Patients with the same genotype have varying genotype incom |
|
|
Incomplete penetrance |
Not all patientswith mutated genotypes show the muted phenotype BRCLA 1 |
|
|
Pleiotropic |
One gene contributes to multiple phenotypic effects |
|
|
Anticipation |
Increase severity or earlier age of onset of a disease in succeeding generation |
|
|
Anticipation |
Increase severity or earlier age of onset of a disease in succeeding generation |
|
|
Loss of heterozygosity |
If a patient inherits or develop a mutation in a tumor suppressor gene the complimentary allele must be deleted/mutated befor cancer develops |
|
|
Dominant negative mutation |
Exerts a dominant effect |
|
|
Linkage disequilibrium |
Tendency for certain alleles at 2 linked loci to occur together more or less often than expected by chance |
|
|
Mosaicism |
Genetically distinct cell lines in the same individual Somatic Gonadal |
|
|
Loci heterogeneity |
Mutation in a differ loci can produce a similar phenotype |
|
|
Loci heterogeneity |
Mutation in a differ loci can produce a similar phenotype |
|
|
Alleles heterogeneity |
Different mutation in the same loci produce similar phenotype |
|
|
Heteroplasmy |
Presence of both normal and mutated mtDNA results in variable expression in mitochondrial inherited disease |
|
|
Uniparental disomy |
Inheritance of 2 copies of a chromosome from 1 parents and no copy form the other parent Heterodisomy- meiosis 1 error Isodisomy- meiosis 2 error |
|
|
McCune Albright syndrome |
Due to Gs-protein activation mutation Unilateral cafe-au-lait spot with ragged edges, polyostotic fibrous dysplasia and at least 1 enodranopaty Lethal if occur before derivation Can service if occurs with mosicism |
|
|
In heterodisomy, a form of uniparental disomy will the inherited alleles be heterozygous or homozygous |
Heterozygous In heterodisomy 2 copies of a chromosome from 1 parent Error in Meiosis 1 |
|
|
Linkage disequilibrium measures what type of group |
A population rather than a family |
|
|
How do most occurrences of uniparental disomy manifest phenotypocally |
A normal phenotype |
|
|
What are 3 malignancies that are commonly associated with loss of heterozygosity |
Retinoblastoma Lifraumeni syndrome Lynch syndrome |
|
|
What term is used when a mitotic error mutation occurs after fertilization and presents in different ways in the same individual |
Somatic mosaicism |
|
|
What is the cause of isodisomy |
Error in misosis II |
|
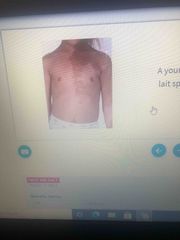
A young girl presents with unilateral cafe au lait spots, polyosototic fibrous dysplasia and precocious pubert |
McCune-Albright syndrome |
|
|
Is the loss of heterozygosity in an oncogene required for cancer to develop |
No, unlike tumor suppressor genes oncogenes do not require a deletion or mutation of the complementary allele |
|
|
What underlying genetic hypothesis best describe the pathogenesis of retinoblastoma |
The 2- hit hypothesis |
|
|
What histological finding would be seen in a bone biopsy specimen on stained from a patient with McCune Albright syndrome |
Bone replaced by collagen and fibroblast Polyostotic fibrous dysplasia |
|
|
Hardy Weinberg equation |
P2 + 2pq + q2 = 1 |
|
|
Hardy Weinberg equation |
P2 + 2pq + q2 = 1 |
|
|
Hardy Weinberg population genetics assumption |
No mutation occurring at the locus No net migration Natural selection is not occurring Large population Completely random mating |
|
|
What does 2pq represent in Hardy Weinberg equilibrium |
Frequency of Heterozygosity |
|
|
What do p2 and q2 represent in Hardy Weinberg population genetics |
P2 - frequency of homozygosity in A q2 frequency of homozygosity in a |
|
|
What is the frequency of an X linked recessive disease in males and females according to hardy Weinberg population genetics |
Males q Females q2 |
|
|
What do p and q represent in Hardy Weinberg population genetics |
P- A q- a |
|
|
Assuming population is in Hardy Weinberg equilibrium the frequency of a dominant allele A is 0.8 what is the frequency of homozygous recessive individual |
0.04 P + q = 1 0.8 + q = 1 q = 0.2 P2 + 2pq + q2 = 1 q2= 0.04 |
|
|
What is Imprinting |
One copy of a gene is silence by methyla only the other copy is expressed |
|
|
Prader willi syndrome |
Material derived gene is silence Disease occur when the paternal allele is deleted or mutated Features hyperplasia, obesity, intellectual disability, hypogondism, hypotonia Chromosome 15 on paternal origin 25% of cases are due to maternal uniparental disomy |
|
|
Angelmans syndrome |
Paternal derived UBE3A gene is silence Disease occur when the maternal allele is deleted or mutated Features seizure, ataxia, severe intellectual disability, inappropriate laughter SAIL UBE3A on chromosome 15 5% of cases due to paternal uniparental disomy |
|
|
Features of prader willi syndrome |
Hyperphagia Obesity Intellectual disability Hypogonadism Hypotonia |
|
|
Features of prader willi syndrome |
Hyperphagia Obesity Intellectual disability Hypogonadism Hypotonia |
|
|
Features of angelman syndrome |
Seizure Ataxia Severe intellectual disability Inappropriate laughter |
|
|
What syndrome results from paternal silencing of UBE3A |
Angelman syndrome |
|
|
What 2 syndromes commonly attributed to genetic imprinting |
Prader willi syndrome Angelman syndrome |
|
|
Autosomal dominant inheritance |
1- Due to defect in structural genes 2- Affect all generations 3- Both male and female affected 4- Often pleotropic and variable expressive 5- Family history is important in the diagnosis 6- If one parent is affected 1/2 is affects |
|
|
Autosomal recessive inheritance |
1- Due to enzyme deficiency 2- Affects 1 generation 3- More sever than dominant 4- 2 carrier heterozygous parent 5- Increase in inbreeding families 6- 1/4 of children affected 1/2 carriers 1/4 unaffected or not a carrier |
|
|
X linked recessive inheritance |
1- 50% of males inherited from heterozygous mothers 2- No male to male transmission 3- skips a generation 4- more sever in males 5- females affected if they homozygous |
|
|
X linked dominant inheritance |
1- transmitted through both parents 2- Heterozygous mother transmit 50% to daughter and son 3- Heterozygous father transmit all to daughters but not son 4- seen in fragile X syndrome, alport syndrome hypophosphetemia rickets (X linked phosphotemia) |
|
|
Hypophosphatemia rickets |
Phosphate wasting a proximal tubule Rickets like presentation- now legs |
|
|
Mitochondrial inheritance |
1- Transmitted only through the mother 2- All offspring of affected females may show signs of disease 3- Variable expression due heteroplasmy |
|
|
Mitochondrial myopathy |
1- Rare 2- Presents with myopathy lactic acidosis CNS features MELAS mitochondrial encephalomyopathy lactic acidosis strike like activity 3- Failure if oxidative phosphorylation 4- Muscle biopsy shoe ragged red edges ( due to diseased mitochondrial in the subsacrolemmia of the muscle) |
|
|
Leber hereditary optic neuropathy |
1- Cel death of the optic nerve neurons 2- Subacute bilateral vision loss in teens and young adults 3- 90% males 4- Permanent |
|
|
What are the signs and symptoms of MELAS syndrome |
Mitochondrial encephalomyopathy Lactic acidosis Strike like activity |
|
|
A boy presents with stroke like activity myopathy and lactic acidosis. His mother has similar symptoms what will muscle biopsy likely show |
Ragged red fibers |
|
|
What modes of inheritance exhibit no father to son transmission |
X linked recessive X linked dominant Mitochondrial inheritance |
|
|
Disorders of which inheritance pattern are often pleiotropic |
Autosomal dominant |
|
|
Offspring in consanguineous families have increased risk of developing disease with what inheritance pattern |
Autosomal recessive |
|
|
What is the chance that 2 heterozygous carriers of autosomal recessive disorder will have an affected offspring |
1/4 25% |
|
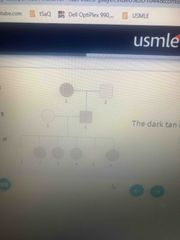
The dark tan color indicated affected individuals Mode of inheritance |
X linked dominant |
|
|
Why are X linked recessive disease more commonly seen in males |
Males only need 1copy of the mutant allele Females need to be homozygous to be affected |
|
|
What is the likelihood that a female carrier of an X linked recessive disease will have an affected son |
50% |
|
|
How does the severity of autosomal recessive disorders compared to that of autosomal dominant disorder |
More severe |
|
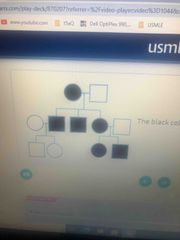
The black color indicates affected individuals Mode of inheritance |
Mitochondrial inheritance |
|
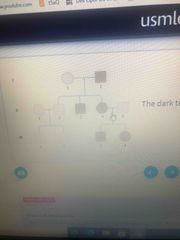
The dark tan color indicated affected individuals Mode of inheritance |
Autosomal dominant |
|
|
What is the probability that an unaffected individual with an affected sibling is a carrier of an autosomal recessive disease |
2/3 |
|

Mode of inheritance |
X linked recessive |
|
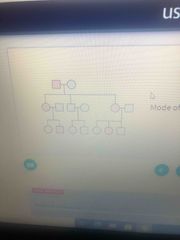
Mode of inheritance |
X linked dominant |
|
|
Autosomal dominant disease |
1- Achondroplasia 2- Autosomal dominant polycystic kidney disease 3- Familial adenomatous polyposis 4- Familial hypercholesterolemib 5- Hereditary hemorrhagic telangiectasia (Osler Weber Randu syndrome) 6- Hereditary spherocytosis 7- Huntington disease 8- Lifraumeni syndrome 9- Marfan syndrome 10- Multiple endocrine neoplasia 11- Myotonic muscular dystrophy 12- Neurofibromatosis type 1 (Von Recklinghausen disease) 13- Neurofibromatosis type 2 14- Tuberoussclerosis 15- Von Hippel Lindau disease |
|
|
Autosomal recessive disease |
1- Autosomal recessive polycystic kidney disease 2- Cystic fibrosis 3- Friedreich ataxia 4- Glycogen storage disease 5- Hemochromatosis 6- Kartagener syndrome 7- Mucopolysaccharidoses (except Hunters disease) 8- Occulocutaneous albinism 9- Phenylketonuria 10- Sickle cell anemia 11- Sphinocyolipidosis 12- Thalassemia 13- Wilson disease |
|
|
Which mucopolysaccharidoses is not autosomal recessive |
Hunters syndrome- Autosomal dominant |
|
|
Which sphinocyolipidosis is not autosomal recessive |
February disease - autosomal dominant |
|
|
Genetics of cystic fibrosis |
1- Autosomal recessive 2- Defect in CFTR gene on Chromosome 7 3- Commonly a deletion of Phe508 4- Most common lethal genetic disorder in Caucasians |
|
|
Genetics of cystic fibrosis |
1- Autosomal recessive 2- Defect in CFTR gene on Chromosome 7 3- Commonly a deletion of Phe508 4- Most common lethal genetic disorder in Caucasians |
|
|
Pathophysiology of cystic fibrosis |
1- CFTR gene encodes for ATP CL channels that secrets CL in the lungs and guts and reabsorb CL in the sweat glands 2- Most common mutation- misfolding of protein- protein retained in RER and not transported to the cell membrane 3- Decrease CL secretion- increase CL in intracellular space results in compensatory increase Na reabsorption by epithelial Na channel 4- Increase H2O reabsorption- abnormally thick mucus to be secreted by gut and lungs 5- Increase Na reabsorption produce a negative tranepitheial potential difference |
|
|
Genetics of cystic fibrosis |
1- Autosomal recessive 2- Defect in CFTR gene on Chromosome 7 3- Commonly a deletion of Phe508 4- Most common lethal genetic disorder in Caucasians |
|
|
Pathophysiology of cystic fibrosis |
1- CFTR gene encodes for ATP CL channels that secrets CL in the lungs and guts and reabsorb CL in the sweat glands 2- Most common mutation- misfolding of protein- protein retained in RER and not transported to the cell membrane 3- Decrease CL secretion- increase CL in intracellular space results in compensatory increase Na reabsorption by epithelial Na channel 4- Increase H2O reabsorption- abnormally thick mucus to be secreted by gut and lungs 5- Increase Na reabsorption produce a negative tranepitheial potential difference |
|
|
Complication of cystic fibrosis |
Pulmonary 1- recurrent pulmonary infection (S.August in children and P. aueydinosa in adults) 2- Allergic bronchopulmonary aspergillosis 3- chronic bronchitis or bronchiectasis (reticularnodular pattern on CXR, opacification of sinuses) 4- nasal polyps GI- 1- Pancreatic insufficiency 2- Malabsorption with steotorrhea 3- Fat soluble vitamin deficiency (ADEK) 4- Biliary cirrhosis 5- liver disease 6- meconium lieus in newborn Reproductive- 1- Infertility in males (absent vas deference) 2- Sub-fertility in females (amenorrhea thick cervical mucus) 3- clubbing of fingers |
|
|
Genetics of cystic fibrosis |
1- Autosomal recessive 2- Defect in CFTR gene on Chromosome 7 3- Commonly a deletion of Phe508 4- Most common lethal genetic disorder in Caucasians |
|
|
Pathophysiology of cystic fibrosis |
1- CFTR gene encodes for ATP CL channels that secrets CL in the lungs and guts and reabsorb CL in the sweat glands 2- Most common mutation- misfolding of protein- protein retained in RER and not transported to the cell membrane 3- Decrease CL secretion- increase CL in intracellular space results in compensatory increase Na reabsorption by epithelial Na channel 4- Increase H2O reabsorption- abnormally thick mucus to be secreted by gut and lungs 5- Increase Na reabsorption produce a negative tranepitheial potential difference |
|
|
Complication of cystic fibrosis |
Pulmonary 1- recurrent pulmonary infection (S.August in children and P. aueydinosa in adults) 2- Allergic bronchopulmonary aspergillosis 3- chronic bronchitis or bronchiectasis (reticularnodular pattern on CXR, opacification of sinuses) 4- nasal polyps GI- 1- Pancreatic insufficiency 2- Malabsorption with steotorrhea 3- Fat soluble vitamin deficiency (ADEK) 4- Biliary cirrhosis 5- liver disease 6- meconium lieus in newborn Reproductive- 1- Infertility in males (absent vas deference) 2- Sub-fertility in females (amenorrhea thick cervical mucus) 3- clubbing of fingers |
|
|
Treatment of cystic fibrosis |
Multifactorial 1- Chest physiotherapy 2- Albuterol 3- Aerosalized for nose (DNase) 4- Hypertonic saline (facilitate mucus clearing) 5- Azithromycine - anti inflammatory 6- ibuprofen- decrease disease progression 7- Pancreatic enzymes 8- In patients with Phe508 deletion lumocaftor (correct misfolding proteins and transport them to cell surface) Ivacalfor (open CL channels) |
|
|
Genetics of cystic fibrosis |
1- Autosomal recessive 2- Defect in CFTR gene on Chromosome 7 3- Commonly a deletion of Phe508 4- Most common lethal genetic disorder in Caucasians |
|
|
Pathophysiology of cystic fibrosis |
1- CFTR gene encodes for ATP CL channels that secrets CL in the lungs and guts and reabsorb CL in the sweat glands 2- Most common mutation- misfolding of protein- protein retained in RER and not transported to the cell membrane 3- Decrease CL secretion- increase CL in intracellular space results in compensatory increase Na reabsorption by epithelial Na channel 4- Increase H2O reabsorption- abnormally thick mucus to be secreted by gut and lungs 5- Increase Na reabsorption produce a negative tranepitheial potential difference |
|
|
Complication of cystic fibrosis |
Pulmonary 1- recurrent pulmonary infection (S.August in children and P. aueydinosa in adults) 2- Allergic bronchopulmonary aspergillosis 3- chronic bronchitis or bronchiectasis (reticularnodular pattern on CXR, opacification of sinuses) 4- nasal polyps GI- 1- Pancreatic insufficiency 2- Malabsorption with steotorrhea 3- Fat soluble vitamin deficiency (ADEK) 4- Biliary cirrhosis 5- liver disease 6- meconium lieus in newborn Reproductive- 1- Infertility in males (absent vas deference) 2- Sub-fertility in females (amenorrhea thick cervical mucus) 3- clubbing of fingers |
|
|
Treatment of cystic fibrosis |
Multifactorial 1- Chest physiotherapy 2- Albuterol 3- Aerosalized for nose (DNase) 4- Hypertonic saline (facilitate mucus clearing) 5- Azithromycine - anti inflammatory 6- ibuprofen- decrease disease progression 7- Pancreatic enzymes 8- In patients with Phe508 deletion lumocaftor (correct misfolding proteins and transport them to cell surface) Ivacalfor (open CL channels) |
|
|
Lumacalfor and Ivacalfor |
Lumacalfor- correct misfolding of proteins transport them to cell surface Ivacalfor- open CL channels |
|
|
What findings may be present on a CT scan of the sinuses in a patient with cystic fibrosis |
Opacification of the sinuses |
|
|
What medication slows the progression of cystic fibrosis |
Ibuprofen |
|
|
What is the benefit of prescribing azithromycin for patients with cystic fibrosis |
Anti inflammatory activity |
|
|
What is the function of aerosolized dornase Alfa, albuterol, inhaled hypertonic saline and chest physiotherapy in the treatment of cystic fibrosis |
Facilities mucus clearance |
|
|
What is the earliest manifestation of cystic fibrosis in a newborn |
Meconium ileus |
|
|
What is the reason for sub fertility in female with cystic fibrosis |
Amenorrhea Thick cervical mucus |
|
|
What is the cause of infertility in male with cystic fibrosis |
Absent vas deference spermatogenesis intact |
|
|
What complications occur in the gastrointestinal tract as a result of pancreatic insufficiency and biliary cirrhosis in patients with cystic fibrosis |
Fat soluble vitamin deficiency (ADEK) Malabsorption with steatorrhea |
|
|
Diagnosis of cystic fibrosis |
1- Increase Cl concentration pilocarpine induce sweat test 2- Contraction alkalosis and hypokalemia 3- Increase immunoreactive trypsinogen in newborns |
|
|
What pathogens most commonly cause pneumonia in infants/children and adults with cystic fibrosis |
Infants/children- S.Aureus Adults- P.Arginosa |
|
|
What findings might be on a chest x Ray from a patient with cystic fibrosis |
Reticulonodular pattern (suggestive bronchiectasis) |
|
|
How does the tranepitheial potential differ with cystic fibrosis |
Increase Na reabsorption causes a negative transepithelial potential difference |
|
|
What does the CFTR gene code for |
ATP gated CL channels to secrets CL in the lungs and gut and reabsorb CL in sweat glands |
|
|
In patients with cystic fibrosis who have Phe508 deletion, what medication function to reduce symptoms by opening chloride channel |
Ivacaftor |
|
|
A patient with known cystic fibrosis have sever malnutrition and steatorrhea what is the treatment |
Pancreatic enzyme replacement |
|
|
Fungi are detected in a lung biopsy specimen obtained from a patient with known cystic fibrosis and recurrent pneumonia diagnosis |
Allergic bronchipulmonary aspergillosis |
|
|
What is the inheritance pattern of cystic fibrosis |
Autosomal recessive |
|
|
X inactivation (lyonization) |
1- One copy of the female X chromosome form a trancriptionally inactive Barr body 2- Female carriers variably affected depending on the pattern of inactivation of the X chromosomes carrying the mutant vs normal gene |
|
|
Why are females with turner syndrome more likely to have an X linked recessive disorder |
They only have 1 X chromosome |
|
|
X linked recessive disorder |
1- Ormithine transcarbamylase deficiency 2- Fabry disease 3- Wiakott- Aldrich syndrome 4- Ocular albinism 5- G6PD deficiency 6- Hunter syndrome 7- Briton agammaglobinemia 8- Hemophilia A and B 9- Lysch Nyhan syndrome 10- Duchenne and Becker muscular dystrophy |
|
|
Duchanes muscular dystrophy |
1- X linked recessive 2- Due to framshift deletion and nonsense mutation 3- Tucated or absent duchane gene 4- Progressive myofiber damage 5- Weakness starts at the pelvic girdle muscle and progress superiority 6- Pseudohypertrophy if the calf muscle- fibrofatty replacement of muscle tissue 7- Waddling gait 8- children < 5 years old 9- Dilated cardiomyopathy common cause of death 10- Gowers sign - use of upper extremity to help to stand up |
|
|
Duchanes muscular dystrophy |
1- X linked recessive 2- Due to framshift deletion and nonsense mutation 3- Tucated or absent duchane gene 4- Progressive myofiber damage 5- Weakness starts at the pelvic girdle muscle and progress superiority 6- Pseudohypertrophy if the calf muscle- fibrofatty replacement of muscle tissue 7- Waddling gait 8- children < 5 years old 9- Dilated cardiomyopathy common cause of death 10- Gowers sign - use of upper extremity to help to stand up |
|
|
Dystrophin gene |
1- Largest protein coded human gene - increase risk of spontaneous mutation 2- Anchors muscle fiber in skeletal and cardiac muscle 3- Connect the intracellular cytoskeleton to the extracellular matrix 4- Loss of dystrophin- myonecrosis 5- Diagnosis increase CK and aldolase |
|
|
Duchanes muscular dystrophy |
1- X linked recessive 2- Due to framshift deletion and nonsense mutation 3- Tucated or absent duchane gene 4- Progressive myofiber damage 5- Weakness starts at the pelvic girdle muscle and progress superiority 6- Pseudohypertrophy if the calf muscle- fibrofatty replacement of muscle tissue 7- Waddling gait 8- children < 5 years old 9- Dilated cardiomyopathy common cause of death 10- Gowers sign - use of upper extremity to help to stand up |
|
|
Dystrophin gene |
1- Largest protein coded human gene - increase risk of spontaneous mutation 2- Anchors muscle fiber in skeletal and cardiac muscle 3- Connect the intracellular cytoskeleton to the extracellular matrix 4- Loss of dystrophin- myonecrosis 5- Diagnosis increase CK and aldolase |
|
|
Becker’s muscular dystrophy |
1- X linked recessive 2- Due to non frameshift mutation in dystrophin gene missense mutation 3- Less sever than duchanes 4- Occur in adolescence and early adulthood |
|
|
Duchanes muscular dystrophy |
1- X linked recessive 2- Due to framshift deletion and nonsense mutation 3- Tucated or absent duchane gene 4- Progressive myofiber damage 5- Weakness starts at the pelvic girdle muscle and progress superiority 6- Pseudohypertrophy if the calf muscle- fibrofatty replacement of muscle tissue 7- Waddling gait 8- children < 5 years old 9- Dilated cardiomyopathy common cause of death 10- Gowers sign - use of upper extremity to help to stand up |
|
|
Dystrophin gene |
1- Largest protein coded human gene - increase risk of spontaneous mutation 2- Anchors muscle fiber in skeletal and cardiac muscle 3- Connect the intracellular cytoskeleton to the extracellular matrix 4- Loss of dystrophin- myonecrosis 5- Diagnosis increase CK and aldolase |
|
|
Becker’s muscular dystrophy |
1- X linked recessive 2- Due to non frameshift mutation in dystrophin gene missense mutation (partially functional instead of truncated) 3- Less sever than duchanes 4- Occur in adolescence and early adulthood |
|
|
Myotonic dystrophy |
1- Autosomal dominant 2- CTG Trinucleotide repeat expansion of DMPK gene 3- Abnormal expression of my tonic protein kinase- myotonia (difficulty releasing hand after handshake) 4- Muscle wasting, cataracts, testicular atrophy, frontal balding, cardiac arrhythmia 5- Have pleiotropic or variable expressivity |
|
|
What is the inheritance pattern of myotonic dystrophy |
Autosomal dominant |
|

The findings in this calf muscle biopsy specimen suggest what diagnosis |
Duchanes muscular dysprrophy |
|

The findings in this calf muscle biopsy specimen suggest what diagnosis |
Duchanes muscular dysprrophy |
|

The findings in this calf muscle biopsy specimen suggest what diagnosis |
Duchanes muscular dystrophy |
|
|
How is the diagnosis of Duchenne muscular dystrophy confirmed |
Genetic testing |
|
|
What 2 enzymes are elevated in patients with Duchenne muscular dystrophy |
Creatine Kinase (CK) Aldolase |
|
|
What’s is the most common cause of death in patients with Duchenne muscular dystrophy |
Dilated cardiomyopathy |
|
|
How does dystrophin connect muscle filaments to the extracellular matrix |
Transmembrane alpha and beta dystroglycan proteins |
|
|
What unique characteristics of the dystrophin gene makes it particularly susceptible to mutation |
The dystrophin gene DMD Largest protein coding human gene |
|
|
In patients with Duchenne muscular dystrophy weakness first presents in which group of muscle before progressing superiorly |
Pelvic girdle muscle |
|
|
A 25 year old male presents with gonadal atrophy early balding cataracts and muscle wasting diagnosis |
Myotonic dystrophy |
|
|
Rhett syndrome |
1- Sporadic disorder 2- Seen in girls (affected males die in uterine or shortly after birth) 3- Due to mutation of MECP2 gene on X chromosome 4- Symptoms occur between 1-4 years old characterized by regression in motor verbal and intellectual ability 5- Ataxia, seizures, growth failure, stereotyped hand wringing, cardiac arrhythmia 6- Life expectancy 40 years old 7- Dies from seizure or cardiac arrhythmia |
|
|
Between what ages does Rett syndrome usually manifest |
1-4 years old |
|
|
What are the clinical manifestation of Rett syndrome |
Ataxia Seizures Growth failure Stereotypic hand wringing Cardiac arrhythmia |
|
|
What is the underlying mutation in Rett syndrome |
MECP2 gene on X chromosome |
|
|
Fragile X syndrome |
1- X linked dominant 2- CGG trinucleotide repeat expansion on FMR1 gene 3- Hypermethylation cause decreasing expression 4- Most common inherited cause of intellectual disability (Down syndrome most common genetic cause) 5- Features 1- post pubertal macroorchidism 2- Long face with Large Jaw 3- Large everted ears 4- Autism 5- Mitral valve prolapse 6- hypermobile joints
|
|
|
What is the most common inherited cause of intellectual disability |
Fragile X syndrome |
|
|
What is the most common inherited cause of intellectual disability |
Fragile X syndrome |
|
|
What is the most common genetic cause of intellectual disability |
Down syndrome |
|
|
What heart defect is most likely present in patients with fragile X syndrome |
Mitral valve prolapse |
|
|
During embryonic development when does the trinucleotide repeat expansion that leads to fragile X syndrome |
Oogenesis |
|
|
What is the mode of inheritance of fragile X syndrome |
X linked dominant |
|
|
How does the trinucleotide repeat expansion in fragile X syndrome affect gene expression |
Hypermethylation Decrease gene expression |
|
|
What are the clinical manifestation of fragile X syndrome |
1- Post pubertal macroorchidism 2- Long face with large jaw 3- Large everted ears 4- Autism 5- Mitral valve prolapse 6- Hypermobile joints |
|
|
Trinucleotide repeat expansion diseases |
Huntington disease Myotonic dystrophy Fragile X syndrome Friedreich ataxia May show genetic anticipation |
|
|
Trinucleotide repeat expansion diseases |
Huntington disease Myotonic dystrophy Fragile X syndrome Friedreich ataxia May show genetic anticipation |
|
|
Trinucleotide repeat expansion in Huntington disease |
CAG AD Caudate decrease in Ach and GABA |
|
|
Trinucleotide repeat expansion diseases |
Huntington disease Myotonic dystrophy Fragile X syndrome Friedreich ataxia May show genetic anticipation |
|
|
Trinucleotide repeat expansion in Huntington disease |
CAG AD Caudate decrease in Ach and GABA |
|
|
Myotonic dystrophy |
CTG AD Cataract, toupee (early balding in men), Gonadal atrophy |
|
|
Trinucleotide repeat expansion diseases |
Huntington disease Myotonic dystrophy Fragile X syndrome Friedreich ataxia May show genetic anticipation |
|
|
Trinucleotide repeat expansion in Huntington disease |
CAG AD Caudate decrease in Ach and GABA |
|
|
Myotonic dystrophy |
CTG AD Cataract, toupee (early balding in men), Gonadal atrophy |
|
|
Fragile X syndrome |
CGG XD Chin Giant Gonads |
|
|
Trinucleotide repeat expansion diseases |
Huntington disease Myotonic dystrophy Fragile X syndrome Friedreich ataxia May show genetic anticipation |
|
|
Trinucleotide repeat expansion in Huntington disease |
CAG AD Caudate decrease in Ach and GABA |
|
|
Myotonic dystrophy |
CTG AD Cataract, toupee (early balding in men), Gonadal atrophy |
|
|
Fragile X syndrome |
CGG XD Chin Giant Gonads |
|
|
Friedreich ataxia |
GAA AR Ataxia GAAit |
|
|
Down syndrome |
1- Incidence 1:700 2- 4% due to unbalanced robertsonian translocation 95% due to meiotic non dysjunction 3- 1% due to post fertilization meiotic error 4- Most common viable chromosomal disorder most common genetic cause of intellectual disability 5- 5 As 1- Advanced maternal age 2- Atresia (duodenal) hirschsprung disease 3- Atrioventricular septal defect 4- Alzheimer’s disease Brushfeild spots 5- AML/ALL 6- Other findings intellectual disability, flat face,prominent epicanthial fold, single Palmer crease, in curved 5th finger, gap between 1st and 2nd toe 7- Increase with advanced maternal age ( <20 1:1500 >45 1:25) 8- First trimester Ultrasound increase nuchal translucency, hyperplastic basal bone 9- Markers increase BHCG and Inhibin A decrease estriol AFP and PAPPA |
|
|
Down syndrome |
1- Incidence 1:700 2- 4% due to unbalanced robertsonian translocation 95% due to meiotic non dysjunction 3- 1% due to post fertilization meiotic error 4- Most common viable chromosomal disorder most common genetic cause of intellectual disability 5- 5 As 1- Advanced maternal age 2- Atresia (duodenal) hirschsprung disease 3- Atrioventricular septal defect 4- Alzheimer’s disease Brushfeild spots 5- AML/ALL 6- Other findings intellectual disability, flat face,prominent epicanthial fold, single Palmer crease, in curved 5th finger, gap between 1st and 2nd toe 7- Increase with advanced maternal age ( <20 1:1500 >45 1:25) 8- First trimester Ultrasound increase nuchal translucency, hyperplastic basal bone 9- Markers increase BHCG and Inhibin A decrease estriol AFP and PAPPA |
|
|
Edward syndrome trisomy 18 |
1- Incidence 1:8000 2- 2nd most common viable chromosomal disorder 3- Findings 1- Prominent occipital 2- Rocker bottom feet 3- Intellectual disability 4- Non- dysjunction 5- Clenched fist with overlapping fingers 6- Low set ears 7- Micronathia 8- Congenital heart defect 9- Omphalocele 10- Myelomingocele 4- Death in the first year of life 5- Markers decrease
|
|
|
Down syndrome |
1- Incidence 1:700 2- 4% due to unbalanced robertsonian translocation 95% due to meiotic non dysjunction 3- 1% due to post fertilization meiotic error 4- Most common viable chromosomal disorder most common genetic cause of intellectual disability 5- 5 As 1- Advanced maternal age 2- Atresia (duodenal) hirschsprung disease 3- Atrioventricular septal defect 4- Alzheimer’s disease Brushfeild spots 5- AML/ALL 6- Other findings intellectual disability, flat face,prominent epicanthial fold, single Palmer crease, in curved 5th finger, gap between 1st and 2nd toe 7- Increase with advanced maternal age ( <20 1:1500 >45 1:25) 8- First trimester Ultrasound increase nuchal translucency, hyperplastic basal bone 9- Markers increase BHCG and Inhibin A decrease estriol AFP and PAPPA |
|
|
Edward syndrome trisomy 18 |
1- Incidence 1:8000 2- 2nd most common viable chromosomal disorder 3- Findings 1- Prominent occipital 2- Rocker bottom feet 3- Intellectual disability 4- Non- dysjunction 5- Clenched fist with overlapping fingers 6- Low set ears 7- Micronathia 8- Congenital heart defect 9- Omphalocele 10- Myelomingocele 4- Death in the first year of life 5- Markers decrease
|
|
|
Patau syndrome trisomy 13 |
1- Incidence 1:15000 2- Defect in fusion of prechordal mesoderm- midline shift 3- Findings 1- Micoencephaly 2- Holoprosencephaly 3- Micro-ophthalmia 4- Cleft lip/palette 5- Congenital heart disease 6- Cutis aplasia 7- Polydactyly 8- Polycystic kidney disease 9- Intellectual disability 10- Rockerbottom feet 4- Death in the first year of life 5- Decrease markers |
|
|
Down syndrome |
1- Incidence 1:700 2- 4% due to unbalanced robertsonian translocation 95% due to meiotic non dysjunction 3- 1% due to post fertilization meiotic error 4- Most common viable chromosomal disorder most common genetic cause of intellectual disability 5- 5 As 1- Advanced maternal age 2- Atresia (duodenal) hirschsprung disease 3- Atrioventricular septal defect 4- Alzheimer’s disease Brushfeild spots 5- AML/ALL 6- Other findings intellectual disability, flat face,prominent epicanthial fold, single Palmer crease, in curved 5th finger, gap between 1st and 2nd toe 7- Increase with advanced maternal age ( <20 1:1500 >45 1:25) 8- First trimester Ultrasound increase nuchal translucency, hyperplastic basal bone 9- Markers increase BHCG and Inhibin A decrease estriol AFP and PAPPA |
|
|
Edward syndrome trisomy 18 |
1- Incidence 1:8000 2- 2nd most common viable chromosomal disorder 3- Findings 1- Prominent occipital 2- Rocker bottom feet 3- Intellectual disability 4- Non- dysjunction 5- Clenched fist with overlapping fingers 6- Low set ears 7- Micronathia 8- Congenital heart defect 9- Omphalocele 10- Myelomingocele 4- Death in the first year of life 5- Markers decrease
|
|
|
Patau syndrome trisomy 13 |
1- Incidence 1:15000 2- Defect in fusion of prechordal mesoderm- midline shift 3- Findings 1- Micoencephaly 2- Holoprosencephaly 3- Micro-ophthalmia 4- Cleft lip/palette 5- Congenital heart disease 6- Cutis aplasia 7- Polydactyly 8- Polycystic kidney disease 9- Intellectual disability 10- Rockerbottom feet 4- Death in the first year of life 5- Decrease markers |
|
|
First trimester marked of trisomies |
BHCG PAPPA |
|
|
Down syndrome |
1- Incidence 1:700 2- 4% due to unbalanced robertsonian translocation between chromosome 14 and 21 95% due to meiotic non dysjunction 3- 1% due to post fertilization meiotic error 4- Most common viable chromosomal disorder most common genetic cause of intellectual disability 5- 5 As 1- Advanced maternal age 2- Atresia (duodenal) hirschsprung disease 3- Atrioventricular septal defect 4- Alzheimer’s disease Brushfeild spots 5- AML/ALL 6- Other findings intellectual disability, flat face,prominent epicanthial fold, single Palmer crease, in curved 5th finger, gap between 1st and 2nd toe 7- Increase with advanced maternal age ( <20 1:1500 >45 1:25) 8- First trimester Ultrasound increase nuchal translucency, hyperplastic basal bone 9- Markers increase BHCG and Inhibin A decrease estriol AFP and PAPPA |
|
|
Edward syndrome trisomy 18 |
1- Incidence 1:8000 2- 2nd most common viable chromosomal disorder 3- Findings 1- Prominent occipital 2- Rocker bottom feet 3- Intellectual disability 4- Non- dysjunction 5- Clenched fist with overlapping fingers 6- Low set ears 7- Micronathia 8- Congenital heart defect 9- Omphalocele 10- Myelomingocele 4- Death in the first year of life 5- Markers decrease
|
|
|
Patau syndrome trisomy 13 |
1- Incidence 1:15000 2- Defect in fusion of prechordal mesoderm- midline shift 3- Findings 1- Micoencephaly 2- Holoprosencephaly 3- Micro-ophthalmia 4- Cleft lip/palette 5- Congenital heart disease 6- Cutis aplasia 7- Polydactyly 8- Polycystic kidney disease 9- Intellectual disability 10- Rockerbottom feet 4- Death in the first year of life 5- Decrease markers |
|
|
First trimester marked of trisomies |
BHCG PAPPA |
|
|
Second trimester markers for trisomies |
BHCG Inhibin Estriol ALP |
|
|
How many normal gametes are produced during gametogenesis of non dysjunction occur during meiosis II |
2 (2 normal, 1 monosomy, 1 trisomy) |
|
|
How many normal gametes are produced during gametogenesis of non dysjunction occur during meiosis I |
0 (2 trisomy, 2 monosomy) |
|
|
Which 2 trisomy disorders may present with severe intellectual disability, rocker bottom feet and congenital heart disease |
Edward syndrome trisomy 18 Patau syndrome trisomy 13 |
|
|
Newborn with Down syndrome does not pass meconium after birth but is not vomiting what GI disease must be considered |
Hirshsprung disease |
|
|
Prenatal screening markers low human chorionic gonadotropin and pregnancy associated plasma protein A diagnosis |
Edward syndrome |
|
|
Prenatal screening markers low BHCG low Inhibin A low estriol low alpha fetoprotein diagnosis |
Edward syndrome |
|
|
Hypoplastic nasal bone and increase nuchal translucency is seen on ultrasound diagnosis |
Down syndrome |
|
|
What is the life expectancy of children born with either trisomy 13 or trisomy 13 |
<1 year |
|
|
What are the prenatyscreen markers for Down syndrome |
Increase BHCG and Inhibin A Decrease PAPPA, Estriol and AFP |
|
|
What is the most common viable autosomal trisomy disorder |
Down syndrome |
|
|
A newborn has exam findings of a single palmar crease, flat facies, and prominent epicanthic folds diagnosis |
Down syndrome |
|
|
Impairment of what embryologic process results in the midline defect seen in patau syndrome |
Defect in fusion of prechordal mesoderm |
|
|
What hematological malignancy are associated with Down syndrome |
Acute myelogenous leukemia Acute lymphoblastic leukemia |
|
|
What opthalmologic finding may be present in a patient with Down syndrome |
Brushfield spots |
|
|
What are the 5 A’s of Down syndrome |
Advanced maternal age Atresia duodenal Atrioventricular septal defect Alzheimer’s disease AML/ALL |
|
|
Signs and symptoms of Edward disease |
Prominent occiput Rocker bottom feet Intellectual disability Non dysjunction Clenched fist with overlapping fingers Low set ears Miconathia Congenital heart defect Omphalocele Myleomenigocele |
|
|
Why early onset Alzheimer’s in Down syndrome |
Chromosome 21 codes for amyloid precursor proteins |
|
|
Robertsonian translocation |
1- Chromosomal translocation that commonly involve chromosome pairs 21, 22, 13, 14 and 15 2- Most common type of translocation 3- Occurs when the 2 long arm of an acrocentric chromosome (chromosome with the centromere near their end) fuse at the centromer 4- 2 short arms are lost 5- Balanced translocation do not cause abnormal phenotype 6- Unbalanced translocation results in miscarriage stillbirth and chromosomal imbalance |
|
|
What feature of chromosome 22, 22, 13, 14 and 15 predisposed them to robertsonian translocation |
Acrocentric chromosomes (chromosomes with their centromer near their end) |
|
|
Cri- du- chat syndrome |
1- Congenital deletion on the short arm of chromosome 5 2- Findings 1- Microphelaphy 2- Moderate to severe intellectual disability 3- High pitched crying/meowing 4- Epicanthal folds 5- Cardiac abnormalities VSD |
|
|
Williams syndrome |
1- Congenital microdeletion of long arm of chromosome 7 (deleted region include elastin gene) 2- Findings 1- Elf facies( long mouth and philtrum) 2- intellectual disability 3- hypercalcemia 4- Well developed verbal skills 5- Extreme friendliness with strangers 6- cardiac abnormalities (supravalvular aortic stenosis, renal artery stenosis) |
|
|
What connective tissue gene is deleted in williams syndrome |
Elastin |
|
|
Name 2 cardiovascular conditions that may be present in patients with williams syndrome |
Supravalvular aortic stenosis Renal artery stenosis |