Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
61 Cards in this Set
- Front
- Back
|
What are the lysosomal storage diseases? General characteristics?
|
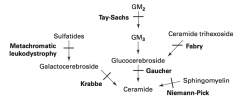
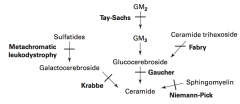
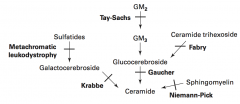
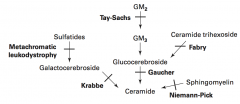
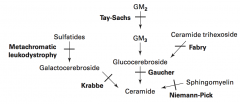
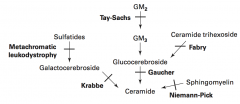
Sphingolipidoses:
- Fabry Disease - Gaucher Disease - Niemann-Pick Disease - Tay-Sachs Disease - Krabbe Disease - Metachromatic Leukodystrophy Mucopolysaccharidoses: - Hurler Syndrome - Hunter Syndrome |
|
|
What is the cause of Fabry disease (lysosomal storage disease)? Findings? What accumulates?
|
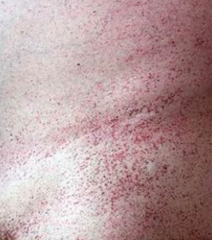
Cause:
- α-Galactosidase A deficiency - X-linked recessive Findings: - Peripheral neuropathy of hands/feet - Angiokeratomas (benign cutaneous lesion of capillaries, resulting in small marks of red to blue color) - CV / Renal disease Accumulation: - Ceramide Trihexoside |
|
|
What is the cause of Gaucher disease (lysosomal storage disease)? Findings? What accumulates? Treatment?
|
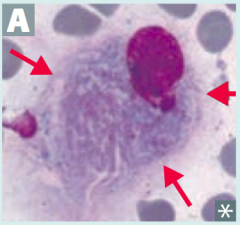
Cause:
- Glucocerebrosidase (β-glucosidase) deficiency - Autosomal recessive Findings: - Most common - Hepatosplenomegaly - Pancytopenia - Aseptic necrosis of femur and bone crises - Gaucher cells (picture) - lipid laden macrophages resembling crumpled tissue paper Accumulation: - Glucocerebroside Treatment: - Recombinant Glucocerebrosidase |
|
|
What is the cause of Niemann-Pick disease (lysosomal storage disease)? Findings? What accumulates?
|
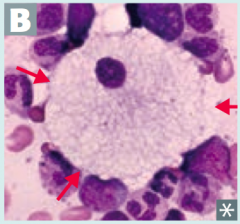
Cause:
- Sphingomyelinase deficiency - Autosomal Recessive Findings: - Progressive neurodegeneration - Hepatosplenomegaly - "Cherry red" spot on macula - Foam cells (lipid-laden macrophages) - picture Accumulation: - Sphingomyelin *No Man Picks (Niemann Pick) his nose with his SPHINGer (sphingomyelinase deficiency)* |
|
|
What is the cause of Tay-Sachs disease (lysosomal storage disease)? Findings? What accumulates?
|
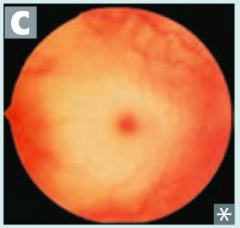
Cause:
- Hexosaminidase A deficiency - Autosomal Recessive Findings: - Progressive neurodegeneration - Developmental delay - "Cherry red" spot on macula - Lysosomes with onion skin - No Hepatosplenomegaly (vs Niemann Pick) Accumulation: - GM2 Ganglioside *Tay-SaX lacks heXosaminidase* |
|
|
What is the cause of Krabbe disease (lysosomal storage disease)? Findings? What accumulates?
|
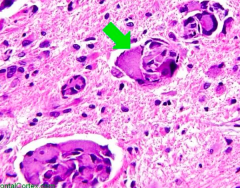
Cause:
- Galactocerebrosidase deficiency - Autosomal recessive Findings: - Peripheral neuropathy - Developmental delay - Optic atrophy - Globoid cells - picture Accumulation: - Galactocerebroside - Psychosine |
|
|
What is the cause of Metachromatic Leukodystrophy (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- Arylsulfatase A deficiency - Autosomal recessive Findings: - Central and peripheral demyelination - Ataxia - Dementia Accumulation: - Cerebroside sulfate |
|
|
What is the cause of Hurler Syndrome (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- α-L-iduronidase deficiency - Autosomal Recessive Findings: - Developmental delay - Gargoylism - Airway obstruction - Corneal clouding - Hepatosplenomegaly Accumulation: - Heparan sulfate - Dermatan sulfate |
|
|
What is the cause of Hunter Syndrome (lysosomal storage disease)? Findings? What accumulates?
|
Cause:
- Iduronate Sulfatase deficiency - X-linked Recessive Findings: - Mild Hurler Syndrome + aggressive behavior - No corneal clouding Accumulation: - Heparan sulfate - Dermatan sulfate *Hunters see clearly (no corneal clouding) and aggressively aim for the X (X-linked recessive* |
|
|
Which lysosomal storage disorders are increased in Ashkenazi Jews?
|
- Tay-Sachs
- Niemann-Pick - Some forms of Gaucher disease |
|
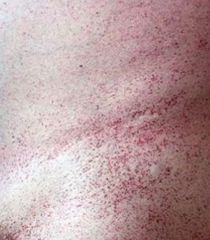
Which disease presents with peripheral neuropathy of hands/feet, angiokeratomas (picture), and CV/renal disease? Cause?
|
Fabry Disease
- Deficiency of α-Galactosidase A - Accumulate Ceramide Trihexoside - X-linked recessive |
|
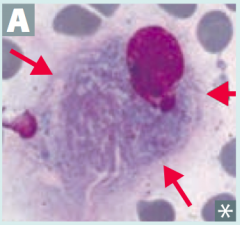
What is the most common lysosomal storage disorder, causing hepatosplenomegaly, pancytopenia, aseptic necrosis of femur, bone crises, and lipid-laden macrophages (picture)? Cause?
|
Gaucher Disease
- Deficiency of Glucocerebrosidase (β-glucosidase) - Accumulate Glucocerbroside - Autosomal recessive |
|
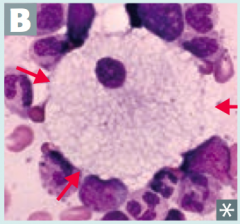
Which disease presents with progressive neurodgeneration, hepatosplenomegaly, cherry red spot on macula, and foam cells (lipid-laden macrophages (picture)? Cause?
|
Niemann-Pick Disease
- Deficiency of Sphingomyelinase (*No Man Picks (Niemann Pick) his nose with his SPHINGer*) - Accumulation of sphingomyelin - Autosomal recessive |
|
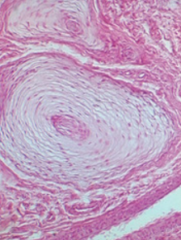
Which disease presents with progressive neurodegeneration, developmental delay, cherry red spot on macula, lysosomes with onion skin (picture), and NO hepatosplenomegaly? Cause?
|
Tay-Sachs Disease
- Deficiency of Hexosaminidase A (*Tay-SaX lacks heXosaminidase*) - Accumulation of GM2 ganglioside - Autosomal recessive |
|
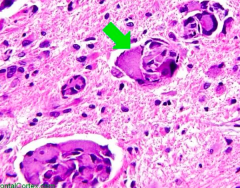
Which disease presents with peripheral neuropathy, developmental delay, optic atrophy, and globoid cell (picture)? Cause?
|
Krabbe Disease
- Deficiency of Galactocerebrosidase - Accumulation of Galactocerebroside and Psychosine - Autosomal Recessive |
|
|
Which disease presents with central and peripheral demyelination with ataxia and dementia? Cause?
|
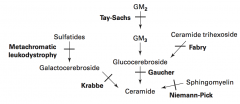
Metachromatic Leukodystrophy
- Deficiency of Arylsulfatase A - Accumulation of Cerebroside Sulfate - Autosomal recessive |
|
|
Which disease presents with developmental delay, gargoylism, airway obstruction, corneal clouding, and hepatosplenomegaly? Cause?
|
Hurler Syndrome
- Deficiency of α-L-iduronidase - Accumulation of heparan sulfate and dermatan sulfate - Autosomal recessive |
|
|
Which disease presents as a mild form of Hurler syndrome with aggressive behavior, but no corneal clouding? Cause?
|
Hunter Syndrome
- Deficiency of iduronate sulfatase - Accumulation of heparan sulfate and dermatan sulfate - X-linked recessive *Hunters see clearly (no corneal clouding) and aggressively aim for the X (X-linked recessive* |
|
|
What is required for fatty acid metabolism / degradation?
|
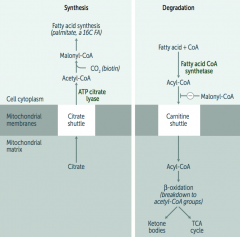
Carnitine-dependent transport into the mitochondrial matrix
|
|
|
What is Carnitine used for? What happens if it is deficient / symptoms?
|
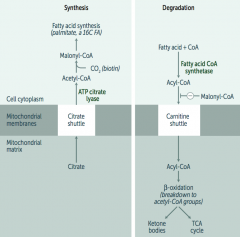
- Carnitine is necessary for long-chain FA degradation
(CARnitine = CARnage of FAs) - Deficiency: inability to transport LCFAs into the mitos, resulting in toxic accumulation; causes weakness, hypotonia, and hypoketotic hypoglycemia |
|
|
What transport shuttle is required for fatty acid synthesis?
|
Citrate Shuttle ("SYtrate" = SYnthesis)
|
|
|
What happens if there is an Acyl-CoA Dehydrogenase deficiency?
|
- ↑ Dicarboxylic Acids
- ↓ Glucose and Ketones - Acetyl-CoA is a (+) allosteric regulator of pyruvate carboxylase in gluconeogenesis - ↓ Acetyl-CoA → ↓ fasting glucose |
|
|
What causes formation of Ketone Bodies?
|
- In prolonged starvation and diabetic ketoacidosis, oxaloacetate is depleted for gluconeogenesis
- In alcoholism, excess NADH shunts oxaloacetate to malate * Both processes cause a build-up of Acetyl-CoA, which shunts glucose and FFA toward production of ketone bodies |
|
|
What are the ketone bodies? What are they made from?
|
Ketone Bodies: Acetoacetate + β-Hydroxybutyrate
- To be used in muscle and brain - Made in liver from fatty acids and amino acids |
|
|
What are the signs and symptoms of Ketone Body accumulation?
|
- Breath smells like acetone (fruity color)
- Urine test for ketones does not detect β-hydroxybutyrate |
|
|
What should you think if the patient's breath smells like acetone?
|
Check their urine for ketone bodies
|
|
|
How many calories are in 1g of protein, carbohydrate, fat, and alcohol?
|
- 1g protein = 4 kcal
- 1g carbohydrate = 4 kcal - 1g alcohol = 7 kcal - 1g fat = 9 kcal |
|
|
What are the sources of fuel for exercise? How long do they last? How does overall performance change with different fuel uses?
|
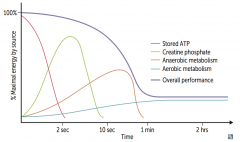
Maximum performance
- <2 seconds: Stored ATP - 2-10 seconds: Creatinine Phosphate Moderate performance - 10 sec - 1 min: Anaerobic Metabolism Low performance - >1 minute: Aerobic Metabolism |
|
|
During fasting and starvation, what are the priorities of the body?
|
- Supply sufficient glucose to the brain and RBCs (RBCs lack mitochondria and so cannot use ketones)
- Preserve protein |
|
|
What happens to metabolism during the fed state (after a meal)?
|
- Glycolysis and aerobic respiration
- Insulin stimulates storage of lipids, proteins, and glycogen |
|
|
What happens to metabolism during the fasting state (between meals)?
|
- Hepatic glycogenolysis (major)
- Hepatic gluconeogenesis and adipose release of FFAs (minor) - Glucagon and adrenaline stimulate use of fuel reserves |
|
|
What happens to metabolism after 1-3 days of starvation?
|
Blood glucose levels maintained by:
- Hepatic glycogenolysis - Adipose release of FFA - Muscle and liver shift fuel use from glucose to FFA - Hepatic gluconeogenesis from peripheral tissue lactate and alanine, and from adipose tissue glycerol and prionyl-CoA (from odd-chain FFA - the only TAG components that contribute to gluconeogenesis) Glycogen reserves are depleted after 1 day RBCs lack mitochondria and so cannot use ketones |
|
|
What happens in the liver after 1-3 days of starvation?
|
- Hepatic glycogenolysis
- Shifts fuel use from glucose to FFAs - Hepatic gluconeogenesis from peripheral tissue lactate and alanine, and from adipose tissue glycerol and propionyl-CoA (from odd-chain FFA) |
|
|
What happens in the adipose after 1-3 days of starvation?
|
- Release of FFA
- FFA used in liver for gluconeogenesis |
|
|
What happens in the muscle after 1-3 days of starvation?
|
- Shifts fuel use from glucose to FFA
- Lactate and alanine are sent to liver for gluconeogenesis |
|
|
What happens to metabolism after 3 days of starvation?
|
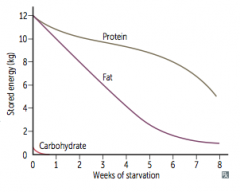
- Adipose stores form ketone bodies - main source of energy for brain
- After depletion of adipose stores, vital protein degradation accelerates, leading to organ failure and death - Amount of excess stores determines survival time |
|
|
What is the rate-limiting step in cholesterol synthesis? Regulation?
|
HMG-CoA Reductase: converts HMG-CoA to Mevalonate
- Induced by insulin - Inhibited by Statin drugs (eg, Lovastatin) competitively and reversibly |
|
|
What happens to plasma cholesterol?
|
2/3 is esterified by lecithin-cholesterol acyltranferase (LCAT)
|
|
|
What enzymes degrade lipids?
|
- Pancreatic Lipase: degrades dietary TGs in small intestine
- Lipoprotein Lipase (LPL): degrades TGs circulating in chylomicrons and VLDLs (found on vascular endothelial surface) - Hepatic TG Lipase (HL): degrades TG remaining in IDL - Hormon-Sensitive Lipase: degrades TG stored in adipocytes |
|
|
Which enzyme degrades dietary TGs in small intestine?
|
Pancreatic Lipase
|
|
|
Which enzyme degrades TG circulating in chylomicrons and VLDLs? Where is it found?
|
Lipoprotein Lipase (LPL) - found on vascular endothelial surface
|
|
|
Which enzyme degrades TG remaining in IDL?
|
Hepatic TG Lipase (HG)
|
|
|
Which enzyme degrades TG stored in adipocytes?
|
Hormone-Sensitive Lipase
|
|
|
What is the action of LCAT (Lecithin-Cholesterol Acyl-Transferase)?
|
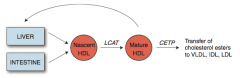
Catalyzes esterification of cholesterol
Converts nascent HDL to mature HDL |
|
|
What is the action of CETP?
|
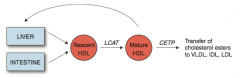
Cholesterol Ester Transfer Protein
- Mediates transfer of cholesterol esters to other lipoprotein particles |
|
|
What are the functions of lipoproteins?
|
- Lipoproteins are composed of varying proportions of cholesterol, TGs, and phospholipids
- LDL and HDL carry most cholesterol - LDL transports cholesterol from liver to tissues (LDL = Lousy) - HDL transports cholesterol from periphery to liver (HDL = Healthy) |
|
|
What is the function of Chylmicrons? Source?
|
- Delivers dietary TGs to peripheral tissue
- Delivers cholesterol to liver in the form of chylomicron remnants, which are mostly depleted for the TAGs - Secreted by intestinal epithelial cells |
|
|
What is the function of VLDL? Source?
|
- Delivers hepatic TGs to peripheral tissue
- Secreted by liver |
|
|
What is the function of IDL? Source?
|
- Formed in the degradation of VLDL
- Delivers TGs and cholesterol to liver |
|
|
What is the function of LDL? Source?
|
- Delivers hepatic cholesterol to peripheral tissues
- Formed by hepatic lipase modification of IDL in the peripheral tissue - Taken up by target cells via receptor-mediated endocytosis |
|
|
What is the function of HDL? Source?
|
- Mediates reverse cholesterol transport from periphery to liver
- Acts as a repository for apoC and apoE (which are needed for chylomicron and VLDL metabolism) - Secreted from both liver and intestine - Alcohol increases synthesis |
|
|
What are the types of familial dyslipidemias?
|
- I: hyperchylmicronemia
- IIa: familial hypercholesterolemia - IV: hyper-triglyceridemia |
|
|
What happens in type I familial dyslipidemia? Cause?
|
Hyperchylmicronemia
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) Cause: autosomal recessive deficiency of lipoprotein lipase or altered apolipoprotein C-II |
|
|
What happens in type IIa familial dyslipidemia? Cause?
|
Familial Hypercholesterolemia
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus Cause: autosomal dominant absent or defective LDL receptors |
|
|
What happens in type IV familial dyslipidemia? Cause?
|
Hyper-Triglyceridemia
- Elevated in blood: VLDL and TG - Pancreatitis Cause: autosomal dominant, hepatic overproduction of VLDL |
|
|
Which disease is caused by lipoprotein lipase deficiency or an altered apolipoprotein C-II?
|
Hyperchylmicronemia / Type I familial dyslipidemia:
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) (Autosomal Recessive) |
|
|
Which disease is caused by absent or defective LDL receptors?
|
Familial Hypercholesterolemia / type IIa familial dyslipidemia:
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus (Autosomal Dominant) |
|
|
Which disease is caused by hepatic overproduction of VLDL?
|
Hyper-Triglyceridemia / type IV Familial Dyslipidemia
- Elevated in blood: VLDL and TG - Pancreatitis (Autosomal Dominant) |
|
|
Which disease is characterized by pancreatitis, hepatosplenomegaly, and eruptive pruritic xanthoams, with no increased risk for atherosclerosis? Cause?
|
Hyperchylmicronemia
- Increased in blood: chylomicrons, TG, and cholesterol - Pancreatitis - Hepatosplenomegaly - Eruptive / prurithic xanthomas (no increased risk for atherosclerosis) Cause: autosomal recessive deficiency of lipoprotein lipase or altered apolipoprotein C-II |
|
|
Which disease is characterized by high cholesterol (~300 mg/dL if heterozygous and ~700+ if homozygous) with accelerated atherosclerosis and possibly MI before age 20; tendon (Achilles) xanthomas and corneal arcus?
|
Familial Hypercholesterolemia
- Increased in blood: LDL and cholesterol - Heterozygotes have cholesterol ~ 300 mg/dL - Homozygotes have cholesterol ~ 700+ mg/dL - Causes accelerated atherosclerosis (may have MI before age 20) - Tendon (Achilles) xanthomas - Corneal arcus Cause: autosomal dominant absent or defective LDL receptors |
|
|
Which disease is characterized by hepatic overproduction of VLDL and causes pancreatitis?
|
Hyper-Triglyceridemia
- Elevated in blood: VLDL and TG - Pancreatitis Cause: autosomal dominant, hepatic overproduction of VLDL |