Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
57 Cards in this Set
- Front
- Back
|
What are the disorders of Galactose Metabolism?
|

- Galactokinase deficiency
- Classic galactosemia |
|
|
What happens in Galactokinase Deficiency? Cause?
|
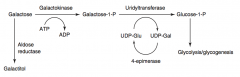
- Hereditary deficiency of Galactokinase
- Autosomal recessive - Galacitol accumulates if galactose present in diet - Relatively mild condition - Galactose appears in blood and urine - Infantile cataracts - May initially present as failure to track objects or to develop a social smile |
|
|
What happens in Classic Galactosemia? Cause? Treat?
|
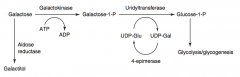
- Absence of Galactose-1-Phosphate Uridyltransferase
- Autosomal Recessive - Damage is caused by accumulation of toxic substances (including galactitol, which accumulates in lens of eye) - Symptoms: failure to thrive, jaundice, hepatomegaly, infantile cataracts, intellectual disability - May lead to E. coli sepsis in neonates - Treatment: exclude galactose and lactose (galactose + glucose) from diet |
|
|
What is the difference in causes of Galactokinase Deficiency and Classic Galactosemia?
|
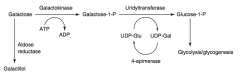
- Galactokinase deficiency: deficiency of galactokinase, galactitol accumulates if galactose present in diet; autosomal recessive
- Classic galactosemia: absence of galactose-1P uridyltransferase, damage is caused by accumulation of toxic substances (galactitol - in lens of eye); depletion of PO4-; autosomal recessive |
|
|
What is the difference in symptoms of Galactokinase Deficiency and Classic Galactosemia?
|
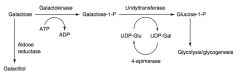
Galactokinase deficiency:
- Mild condition - Galactose appears in blood and urine - Infantile cataracts - may initially present as failure to track objects or to develop a social smile Classic galactosemia: - Failure to thrive - Jaundice - Hepatomegaly - Infantile cataracts (accumulation of galactitol in lens) - Intellectual disability - Can lead to E. coli sepsis in neonates |
|
|
Which of the disorders of fructose deficiency is similar to one of the disorders of galactose deficiency?
|
Fructose Intolerance (d/t deficiency of Aldolase B) is similar to Classic Galactosemia (d/t deficiency of Galactose-1P Uridyltransferase
*FAB-GUT* Fructose is to Aldolase B as Galactose is to Uridyl-Transferase |
|
|
What is an alternative method of trapping glucose in the cell?
|

Convert it to its alcohol counterpart, called Sorbitol, via Aldose Reductase
|
|
|
How is sorbitol synthesized? What can happen to it next?
|

1. Glucose → Sorbitol (via Aldose Reductase and NADPH)
2. Sorbitol → Fructose (via Sorbitol Dehydrogenase and NAD+) 2nd reaction only occurs in certain tissues (liver, ovaries, seminal vesicles) |
|
|
What are the implications if tissues don't have both enzymes to generate sorbitol and to dehydrogenate sorbitol?
|

- Tissues like Schwann cells, retina, the lens, and kidneys only have Aldose Reductase to generate Sorbitol
- These tissues are at risk for intracellular sorbitol accumulation, causing osmotic damage (eg, cataracts, retinopathy, and peripheral neuropathy with chronic hyperglycemia in diabetes) |
|
|
What causes cataracts, retinopathy, and peripheral neuropathy in patients with chronic hyperglycemia / diabetes?
|

- Glucose is converted to Sorbitol via Aldose Reductase
- Schwann cells (→ peripheral neuropathy), retina (→ retinopathy), lens (→ cataracts), and kidneys don't have sufficient Sorbitol Dehydrogenase to remove Sorbitol, leading to OSMOTIC DAMAGE |
|
|
Besides glucose, high levels of what other molecule can also be converted by Aldose Reductase? Product?
|
Galactose → Galactitol (via Aldose Reductase)
|
|
|
What causes Lactose Intolerance?
|
- Insufficient lactase enzyme → dietary lactose intolerance
- Lactase functions on the brush border to digest lactose (in human and cow milk) into glucose and galactose |
|
|
What are the two types of lactase deficiency? How do they differ?
|
Primary:
- Age dependent decline after childhood (Absence of lactase-persistent allele) - Common in Asian, African, or Native American heritage Secondary: - Loss of brush border d/t gastroenteritis (eg, rotavirus), autoimmune disease, etc |
|
|
What are the signs / symptoms of Lactose Intolerance?
|
- Stool demonstrates ↓ pH
- Breath shows ↑ H+ content with lactose tolerance test - Intestinal biopsy reveals normal mucosa in patients with hereditary lactose intolerance - Bloating, cramps, flatulence, osmotic diarrhea |
|
|
How do you treat Lactose Intolerance?
|
Avoid dairy products or add lactase pills to diet
|
|
|
What form of amino acids are found in proteins in humans?
|
Only L-form of amino acids
|
|
|
What type of amino acids need to be supplied in the diet?
|
Essential Amino Acids:
- Glucogenic: Met, Val, His - Glucogenic / Ketogenic: Ile, Phe, Thr, Trp - Ketogenic: Leu, Lys |
|
|
What are the glucogenic essential amino acids?
|
- Methionine (Met)
- Valine (Val) - Histidine (His) |
|
|
What are the glucogenic / ketogenic essential amino acids?
|
- Isoleucine (Ile)
- Phenylalanine (Phe) - Threonine (Thr) - Tryptophan (Trp) |
|
|
What are the ketogenic essential amino acids?
|
- Leucine (Leu)
- Lysine (Lys) |
|
|
What are the acidic amino acids?
|
- Aspartic Acid (Asp)
- Glutamic Acid (Glu) Negatively charged at body pH |
|
|
What are the basic amino acids?
|
- Arginine (Arg) - most basic
- Lysine (Lys) - Histidine (His) - no charge at body pH |
|
|
Which amino acids are required during periods of growth?
|
- Arginine (Arg) - most basic AA
- Histidine (His) - basic, but no charge at body pH |
|
|
Which amino acids are increased in histones? Function?
|
- Arginine (Arg) - most basic AA
- Lysine (Lys) - basic They bind negatively charged DNA |
|
|
What process is necessary for amino acid catabolism?
|
Urea Cycle
|
|
|
What is the function of the Urea Cycle?
|
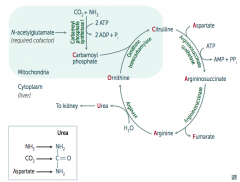
- AA catabolism results in the formation of common metabolites (eg, pyruvates, acetyl-CoA)
- These serve as metabolite fuels - Excess nitrogen (NH3) generated by this process is converted to urea and excreted by the kidneys |
|
|
How do you remember the intermediates in the Urea Cycle?
|
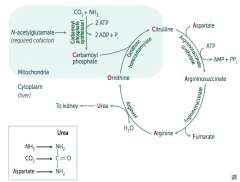
Ordinarily, Careless Crappers Are Also Frivolous About Urination:
- Ornithine + Carbomoyl phosphate → - Citrulline + Aspartate → - Argininosuccinate → - Arginine → (+Fumarate) - Urea→ |
|
|
What are the enzymes in the Urea Cycle?
|
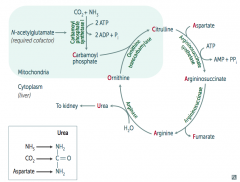
1. Ornithine Transcarbamylase
2. Argininosuccinate Synthetase 3. Argininosuccinase 4. Arginase |
|
|
What is Urea made of?
|
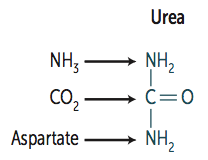
- NH3 (ammonia)
- CO2 - Aspartate (donates NH2) |
|
|
How is Carbamoyl Phosphate synthesized for the Urea Cycle?
|
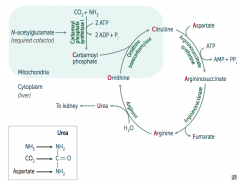
In mitochondria, Carbamoyl Phosphate Synhtetase I combines CO2 + NH3
Uses 2 ATP and requires N-acetylglutamate as a cofactor |
|
|
What happens to Carbamoyl Phosphate in Urea Cycle?
|
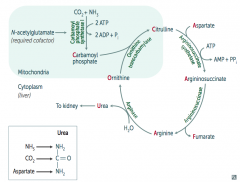
Combines with Ornithine via Ornithine Transcarbamylase → Citrulline
|
|
|
What happens to Citrulline in Urea Cycle?
|
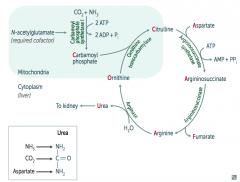
Citrulline combines with Aspartate via Argininosuccinate Synthetase → Argininosuccinate
Requires one ATP → AMP + PPi |
|
|
What happens to Argininosuccinate in Urea Cycle?
|
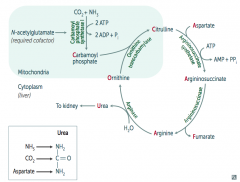
Argininosuccinase breaks it down into Fumarate (released) and Arginine (continues in Urea Cycle)
|
|
|
What happens to Arginine in Urea Cycle?
|
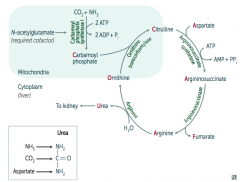
Arginase combines Arginine with H2O to release Urea (which goes to kidney) and Ornithine (regenerated to continue Urea Cycle)
|
|
|
What processes allow safe transport of ammonia in the body?
|
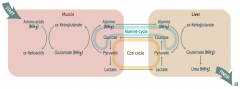
Cori Cycle and Alanine Cycle
|
|
|
How do amino acids (ammonia / NH3) get safely transported from the muscle to the liver (to be converted to urea)?
|
Muscle:
- Amino Acids (NH3) + α-Ketoglutarate → Glutamate (NH3) + α-Ketoacids - Glutamate (NH3) + Pyruvate → Alanine (NH3) + α-Ketoglutarate Alanine Cycle: - Alanine (NH3) transported to liver Liver: - Alanine (NH3) + α-Ketoglutarate → Glutamate (NH3) + Pyruvate - Glutamate (NH3) →→ Urea (NH3) → kidney |
|
|
What is the Alanine Cycle?
|
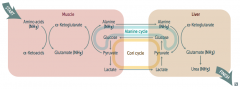
1. Alanine (NH3) transported from muscle to liver
2. Alanine (NH3) + α-Ketoglutarate → Glutamate (NH3) + Pyruvate 3. Pyruvate → Glucose 4. Glucose transported from liver back to muscle 5. Glucose → Pyruvate 6. Pyruvate + Glutamate (NH3) → Alanine (NH3) + α-Ketoglutarate |
|
|
What is the Cori Cycle?
|
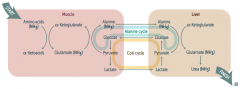
In muscle:
- Glucose → Pyruvate - Pyruvate → Lactate Cori Cycle: - Lactate transported from muscle to liver In liver: - Lactate → Pyruvate - Pyruvate → Glucose (overlaps with Alanine Cycle) Cori / Alanine Cycle: - Glucose transported from liver back to muscle |
|
|
What can cause Hyperammonemia?
|
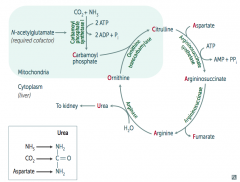
Acquired (eg, liver disease)
Hereditary (eg, urea cycle enzyme deficiency) - N-acetylglutamate deficiency - Ornithine transcarbamylase deficiency - Etc. |
|
|
What does Hyperammonemia cause? Symptoms?
|
- Excess NH4+ → depletes α-Ketoglutarate, leading to inhibition of TCA cycle
- Symptoms: tremor (asterixis), slurring of speech, somnolence, vomiting, cerebral edema, blurring of vision |
|
|
How do you treat Hyperammonemia?
|
- Limit protein in diet
- Benzoate or phenylbutyrate (both of which bind AA and lead to excretion) can be given to ↓ ammonia levels) - Lactulose can acidify the GI tract and trap NH4+ for excretion |
|
|
What are the implications of an N-acetylglutamate deficiency?
|
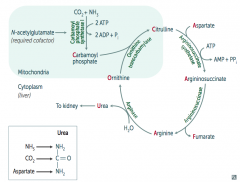
- Required cofactor for Carbamoyl Phosphate Synthetase I
- Absence → Hyperammonemia (because Urea Cycle requires Carbamoyl Phosphate) |
|
|
What is the presentation of an N-acetylglutamate deficiency?
|
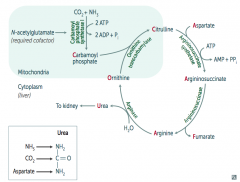
Identical to Carbamoyl Phosphate Synthetase I deficiency:
- Hyperammonemia: tremor (asterixis), slurring of speech, somnolence, vomiting, cerebral edema, blurring of vision - ↑ Ornithine with normal urea cycle enzymes suggests hereditary N-acetylglutamate deficiency |
|
|
What is the most common urea cycle disorder? Cause?
|
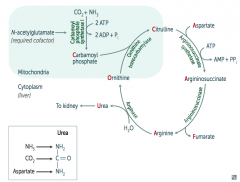
- Ornithine Transcarbamylase Deficiency (which is supposed to combine Carbamoyl Phosphate and Ornithine to make Citrulline)
- X-linked recessive |
|
|
What are the implications of an Ornithine Transcarbamylase Deficiency?
|
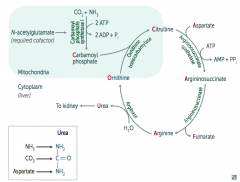
- Interferes with the body's ability to eliminate ammonia
- Often evident in the first few days of life, but may present with late onset - Excess carbamoyl phosphate is converted to orotic acid (part of the pyrimidine synthesis pathway) |
|
|
What are the lab findings of Ornithine Transcarbamylase Deficiency?
|
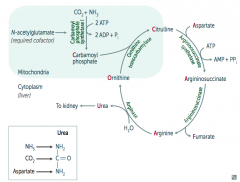
- ↑ Orotic acid in blood and urine (made from excess unused Carbamoyl Phosphate)
- ↓ BUN - Hyperammonemia - No megaloblastic anemia (vs orotic aciduria) |
|
|
What are the amino acid derivatives of Phenylalanine?
|
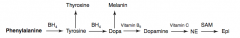
- Tyrosine → Thyroxine
- Melanin - Dopamine → Norepinephrine → Epinephrine |
|
|
What are the amino acid derivatives of Tryptophan?
|
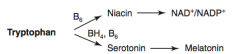
- NAD+ / NADP+
- Serotonin → Melatonin |
|
|
What are the amino acid derivatives of Histidine?
|
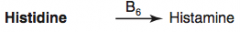
Histamine
|
|
|
What are the amino acid derivatives of Glycine?
|

Porphyrin → Heme
|
|
|
What are the amino acid derivatives of Glutamate?
|
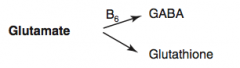
- GABA
- Glutathione |
|
|
What are the amino acid derivatives of Arginine?
|
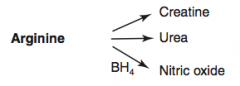
- Creatine
- Urea - Nitric Oxide |
|
|
How is Thyroxine (T4) synthesized?
|
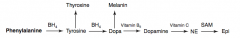
Phenyalanine (BH4) → Tyrosine → Thyroxine
|
|
|
How is Melanin synthesized?
|
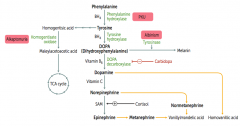
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa → Melanin
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Melanin via Tyrosinase |
|
|
How is Dopamine synthesized?
|
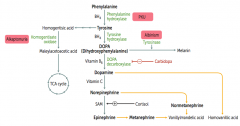
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase |
|
|
How is Norepinephrine synthesized?
|
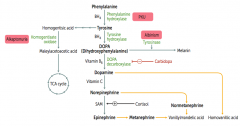
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine (Vitamin C) → Norepinephrine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase Dopamine → NE |
|
|
How is Epinephrine synthesized?
|
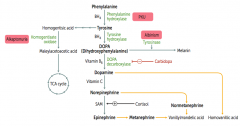
Phenyalanine (BH4) → Tyrosine (BH4) → Dopa (Vitamin B6) → Dopamine (Vitamin C) → Norepinephrine (SAM) → Epinephrine
Phe → Tyr via Phenylalanine Hydroxylase Tyr → DOPA via Tyrosine Hydroxylase DOPA → Dopamine via DOPA Decarboxylase Dopamine → NE requires Vitamin C NE → Epinephrine requires SAM |