Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
54 Cards in this Set
- Front
- Back
|
What reaction is mediated by the Pyruvate Dehydrogenase Complex?
|
Pyruvate + NAD+ + CoA →
Acetyl-CoA + CO2 + NADH |
|
|
What are the requirements of the Pyruvate Dehydrogenase Complex? How many enzymes are part of the complex?
|
Contains 3 enzymes that require 5 cofactors:
1) Pyrophosphate (B1, thiamine; TPP) 2) FAD (B2, riboflavin) 3) NAD (B3, niacin) 4) CoA (B5, pantothenic acid) 5) Lipoic Acid |
|
|
What activates the Pyruvate Dehydrogenase Complex?
|
- ↑ NAD+/NADH ratio
- ↑ ADP - ↑ Ca2+ |
|
|
What is the Pyruvate Dehydrogenase Complex similar to?
|
α-Ketoglutarate Dehydrogenase Complex
- Same cofactors, similar substrate and action - Converts α-Ketoglutarate → Succinyl-CoA (TCA Cycle) |
|
|
Which poison affects the Pyruvate Dehydrogenase Complex? Symptoms?
|
Arsenic: inhibits lipoic acid which is a necessary cofactor for the complex
Symptoms: - Vomiting - Rice water stools - Garlic breath |
|
|
What are the implications of a Pyruvate Dehydrogenase Complex Deficiency?
|
Causes backup of substrate (pyruvate and alanine) leading to LACTIC ACIDOSIS
|
|
|
What is the most common cause of a Pyruvate Dehydrogenase Complex deficiency?
|
Mutations in X-linked gene for E1-α subunit of PDC
|
|
|
What are the findings of a Pyruvate Dehydrogenase Complex deficiency (most cases d/t mutations in X-linked gene for E1-α subunit of PDC)? How do you treat?
|
Symptoms:
- Neurologic defects, usually starting in infancy Treatment: - ↑ Intake of ketogenic nutrients (eg, high fat content or ↑ lysine and leucine) - Lysine and leucine are the onLy pureLy ketogenic amino acids |
|
|
What are the possible pyruvate metabolic pathways
|
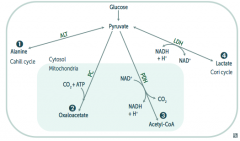
1. Pyruvate Dehydrogenase Complex: transition from glycolysis to TCA cycle
2. Alanine Aminotransferase 3. Pyruvate Carboxylase 4. Lactic Acid Dehydrogenase |
|
|
What enzyme(s) shuttle pyruvate into the TCA cycle? Cofactors?
|
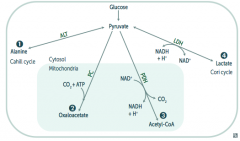
Pyruvate Dehydrogenase Complex
- Pyruvate → Acetyl-CoA - B1 - Pyrophosphate - B2 - FAD - B3 - NAD - B5 - CoA - Lipolic Acid |
|
|
What enzyme(s) converts pyruvate into a form that can be carried from the liver to muscle? Cofactors?
|
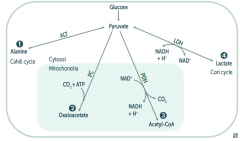
Alanine Aminotransferase
- Pyruvate → Alanine (alanine carries amino groups to the liver from muscle) - B6 - Pyridoxine |
|
|
What enzyme(s) converts pyruvate into a form that can replenish the TCA cycle or be used in gluconeogenesis? Cofactors?
|
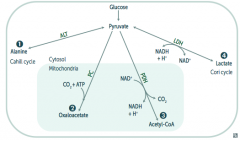
Pyruvate Carboxylase
- Pyruvate → Oxaloacetate (via addition of CO2 + ATP) - B7 - Biotin |
|
|
What enzyme(s) converts pyruvate into a form that can enter the Cori cycle? Cofactors?
|
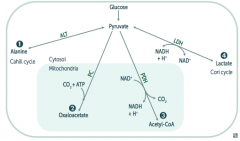
Lactic Acid Dehydrogenase
- Pyruvate → Lactate (enters Cori cycle) - B3 - NAD |
|
|
In what tissues is Lactic Acid Dehydrogenase conversion of Pyruvate to Lactate the major pathway?
|
- RBCs
- Leukocytes - Kidney medulla - Lens - Testes - Cornea |
|
|
What is the effect / results of the TCA Cycle?
|
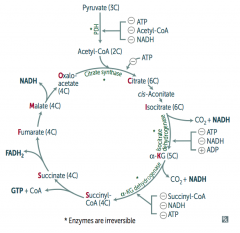
Pyruvate → Acetyl-CoA produces 1 NADH, 2 CO2
For every Acetyl-CoA - 3 NADH - 1 FADH2 - 2 CO2 - 1 GTP - 10 ATP (2x everything per glucose) |
|
|
How do you remember the substrates / intermediates of the Kreb's Cycle?
|
Citrate is Kreb's Starting Substrate For Making Oxaloacetate
- Citrate (6C) - Isocitrate (6C) - α-Ketoglutarate (5C) + CO2 - Succinyl-CoA (4C) + CO2 - Succinate (4C) - Fumarate (4C) - Malate (4C) - Oxaloacetate (4C) + Acetyl-CoA (2C) |
|
|
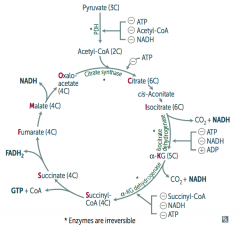
What regulates the TCA cycle?
|
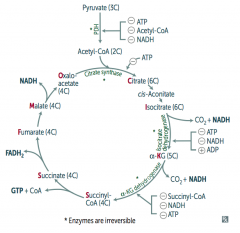
Pyruvate Dehydrogenase Complex
- ATP (-) - Acetyl-CoA (-) - NADH (-) Citrate Synthase (Oxaloacetate + Acetyl-Coa → Citrate) - ATP (-) Isocitrate Dehydrogenase (Isocitrate → α-Ketoglutarate + CO2) - ATP (-) - NADH (-) - ADP (+) α-Ketoglutarate Dehydrogenase (α-KG → Succinyl-Coa + CO2) - Succinyl-CoA (-) - NADH (-) - ATP (-) |
|
|
Which enzymes in the TCA cycle are irreversible?
|
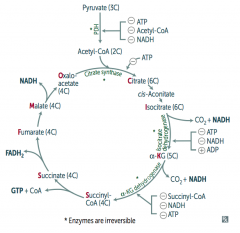
The same steps that were regulated by ATP, etc)
- Pyruvate Dehydrogenase Complex - Citrate Synthase - Isocitrate Dehydrogenase - α-Ketoglutarate Dehydrogenase |
|
|
What happens in the Electron Transport Chain and Oxidative Phosphorylation?
|
- NADH electrons from glycolysis enter mitochondria via malate-aspartate or glycerol-3-phosphate shuttle
- FADH2 electrons are transferred to complex II (at lower energy level than NADH) - Passage of e- results in formation of a H+ gradient that, coupled to oxidative phosphorylation, drives production of ATP |
|
|
How does NADH enter the Electron Transport Chain?
|
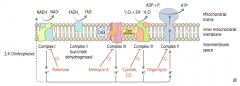
NADH electrons from glycolysis enter mitochondria via malate-aspartate or glycerol-3-phosphate shuttle
|
|
|
How does FADH2 enter the Electron Transport Chain?
|
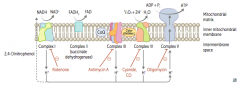
FADH2 electrons are transferred to complex II (at lower energy level than NADH)
|
|
|
How many ATP are produced per NADH and FADH2 through the Electron Transport Chain and Oxidative Phosphorylation?
|
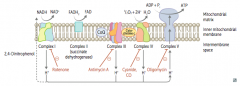
1 NADH → 2.5 ATP (remember, niacin is vitamin B3)
1 FADH2 → 1.5 ATP (remember, flavin is vitamin B2) |
|
|
What drugs / poisons can affect the Electron Transport Chain and Oxidative Phosphorylation?
|
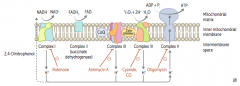
- Electron Transport Inhibitors: Rotenone, Cyanide, Antimycin A, CO
- ATP Synthase Inhibitors: Oligomycin - Uncoupling Agents: 2,4-DNP, Aspirin, Thermogenin (in brown fat) |
|
|
What is the effect of electron transport inhibitors? Which drugs?
|
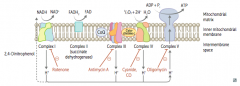
- Directly inhibits electron transport, causing a ↓ proton gradient and block of ATP synthesis
- Rotenone: inhibits Complex I - Antimycin A: inhibits Complex III - Cyanide and CO: inhibit Complex IV |
|
|
What is the effect of ATP Synthase Inhibitors? Which drugs?
|
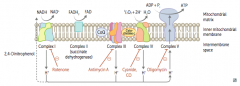
- Directly inhibit mitochondrial ATP synthase, causing an ↑ proton gradient
- No ATP is produced because electron transport stops - Oligomycin: inhibits Complex V (ADP + Pi → ATP + H2O) |
|
|
What is the effect of Uncoupling Agents? Which drugs?
|
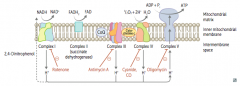
- ↑ Permeability of membrane, causing a ↓ proton gradient and ↑ O2 consumption
- ATP synthesis stops, but electron transport continues - PRODUCES HEAT - 2,4-DNP - Aspirin (fevers often occur after aspirin overdose) - Thermogenin in brown fat |
|
|
What are the irreversible enzymes in Gluconeogenesis?
|
Pathway Produces Fresh Glucose
- Pyruvate carboxylase - PEP carboxykinase - Fructose-1,6-bisphosphatase - Glucose-6-phosphatase |
|
|
What is the action of Pyruvate Carboxylase? Requirements? Location?
|
- Irreversible conversion of Pyruvate → Oxaloacetate
- Requires biotin and ATP - Activated by acetyl-CoA - In mitochondria |
|
|
What is the action of PEP Carboxykinase? Requirements? Location?
|
- Irreversible conversion of Oxaloacetate → Phosphoenolpyruvate (PEP)
- Requires GTP - In cytosol |
|
|
What is the action of Fructose-1,6-Bisphosphatase? Location?
|
- Irreversible conversion of Fructose-1,6-Bisphosphate → Fructose-6P
- In cytosol |
|
|
What is the action of Glucose-6-Phosphatase? Location?
|
- Irreversible conversion of Glucose-6-Phosphate → Glucose
- In ER |
|
|
Where does gluconeogenesis occur? Where can't it occur?
|
* Primarily in the liver
- Enzymes also found in kidney and intestinal epithelium - Muscle cannot participate in gluconeogenesis because it lacks glucose-6P |
|
|
What are the implications of a deficiency of the gluconeogenic enzymes?
|
Hypoglycemia
|
|
|
Which fatty acids can be used for gluconeogenesis? Why/why not?
|
- Odd-chain FA yield 1 propionyl-CoA during metabolism, which can enter the TCA cycle as succinyl-CoA, undergo gluconeogenesis, and serve as a glucose source
- Even-chain FA can't produce new glucose, since they yield only acetyl-CoA equivalents |
|
|
What is the function of the HMP shunt (Pentose Phosphate Pathway)?
|
- Provides a source of NADPH from an abundantly available glucose-6P
- NADPH is required for reductive reactions (eg, glutathione reduction inside RBCs) - Also yields Ribose for nucleotide synthesis and glycolytic intermediates |
|
|
How many ATP are used/gained from HMP shunt (Pentose Phosphate Pathway)?
|
No ATP are used or produced
|
|
|
Where does the HMP shunt (Pentose Phosphate Pathway) take place?
|
- Lactating mammary glands
- Liver - Adrenal cortex (sites of FA or steroid synthesis) - RBCs |
|
|
What are the reactions in the HMP shunt (Pentose Phosphate Pathway)?
|
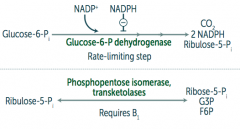
1. Oxidative (irreversible)
Glucose-6P → Ribulose-5P + CO2 + 2 NADPH by Glucose-6P dehydrogenase 2. Non-Oxidative (reversible) Ribulose-5P → Ribose-5P + G3P + F6P by Phosphopentose isomerase and transketolases |
|
|
What is the rate-limiting step of the HMP shunt (Pentose Phosphate Pathway)?
|
Glucose-6P Dehydrogenase (oxidative/irreversible reaction)
- Glucose-6P → Ribulose-5P + CO2 + 2 NADPH |
|
|
What mediates the respiratory / oxidative burst in neutrophils and/or monocytes?
|
Activation of membrane-bound NADPH oxidase (in neutrophils and monocytes)
|
|
|
What is the function of the respiratory / oxidative burst in neutrophils and/or monocytes?
|
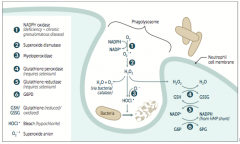
Important in the immune response → rapid release of reactive oxygen intermediates (ROIs)
* NADPH plays a role in the creation of ROIs and in their neutralization |
|
|
What do the WBCs of patients with Chronic Granulomatous Disease do? Implications for further infection?
|
- WBCs can utilize H2O2 generated by invading organisms and convert it to ROIs
- Patients are at ↑ risk for infection by catalase-positive species (eg, S. aureus and Aspergillus) because they neutralize there own H2O2, leaving WBCs without ROIs for fighting infections |
|
|
What is the function of reduced glutathione? How does glutathione get reduced?
|
- Reduced glutathione detoxifies free radicals and preoxides
- NADPH is necessary to keep glutathione reduced |
|
|
What are the implications of reduced availability of NADPH?
|
RBCs:
- Hemolytic anemia d/t poor RBC defense against oxidizing agents (eg, fava beans, sulfonamides, primaquine, anti-TB drugs) - Infection can also precipitate hemolysis (free radicals generated by inflammatory response can diffuse into RBCs and cause oxidative damage) |
|
|
What are the implications of a glucose-6P dehydrogenase deficiency?
|
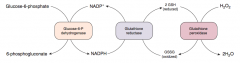
- Glucose 6-P Dehydrogenase is responsible for regenerating NADPH from NADP+
- NADPH is necessary to reduce GSSG (oxidized glutathione) to GSH (reduced glutathione) - GSH is necessary to detoxify free radicals and peroxides |
|
|
What causes glucose-6P dehydrogenase deficiency? Who is more likely to get this?
|
- X-linked recessive disorder
- Most common human enzyme deficiency - More prevalent among blacks as it confers ↑ malarial resistance |
|
|
What does glucose-6P dehydrogenase deficiency cause?
|
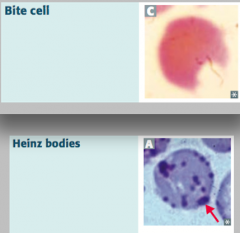
Heinz bodies:
- Oxidized hemoglobin precipitated within RBCs Bite cells: - Results from phagocytic removal of Heinz bodies by splenic macrophages *Think, "Bite into some Heinz ketchup"* |
|
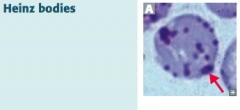
What are Heinz bodies? What are they a sign of?
|
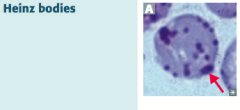
- Oxidized Hemoglobin precipitated within RBCs
- Caused by Glucose-6P Dehydrogenase (G6PD) deficiency (X-linked recessive) |
|

What are bite cells? What are they a sign of?
|
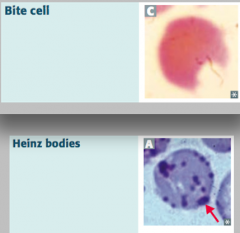
- Result from phagocytic removal of Heinz bodies by splenic macrophages
- Caused by Glucose-6P Dehydrogenase (G6PD) deficiency (X-linked recessive) |
|
|
What are the disorders of fructose metabolism?
|
- Essential fructosuria
- Fructose intolerance |
|
|
What happens in Essential Fructosuria? Cause?
|
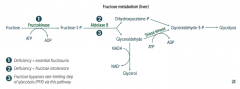
- Defect in Fructokinase (1)
- Autsomal Recessive - Benign, asymptomatic condition, since fructose is not trapped in cells - Fructose appears in blood and urine - Mild condition compared to analogous disorders in galactose metabolism |
|
|
What happens in Fructose Intolerance? Cause? Treatment?
|
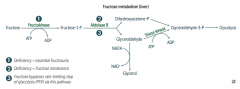
- Hereditary deficiency of Aldolase B (2)
- Autosomal recessive - Fructose-1P accumulates, causing ↓ in available phosophate, which inhibits glycogenolysis and gluconeogenesis - Symptoms occur after consuming fruit, juice, or honey - Urine dipstick will be negative (tests for glucose only); reducing sugar can be detected in urine (nonspecific test for inborn errors of CHO metabolism) - Symptoms: hypoglycemia, jaundice, cirrhosis, vomiting - Tx: ↓ intake of both fructose and sucrose (glucose + fructose) |
|
|
What is the difference in causes of Essential Fructosuria and Fructose Intolerance?
|
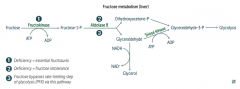
- Essential Fructosuria: defect in Fructokinase, autosomal recessive
- Fructose Intolerance: deficiency of Aldolase B, autosomal recessive; fructose-1P accumulates, causing ↓ availability of phosphate, which inhibits glycogenolysis and gluconeogenesis (less glucose available) |
|
|
What is the difference in symptoms of Essential Fructosuria and Fructose Intolerance?
|
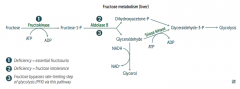
Essential Fructosuria:
- Asymptomatic (fructose is not trapped in cells) - Fructose appears in urine and blood Fructose Intolerance: - Hypoglycemia (d/t inhibition of glycogenolysis and gluconeogenesis) - Jaundice and cirrhosis - Vomiting |