Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
40 Cards in this Set
- Front
- Back
|
*How do Brain tumors differ from Lower Organ Tumors?
|
-They may be histologically benign but biologically malignant (LOCATION LOCATION LOCATION, infiltrate adjacent brain): they are difficult to resect and can compress vital structures
-Staging is not defined (no TMN system because there is no concept of margins, no CNS lymphatic drainage, and rare metastasis) -They may infiltrate adjacent brain: Subpial/subarachnoid spread, Perineuronal spread, Perivascular spread |
|
|
What is the sole proven environmental risk factor for the development of brain tumors?
What is the common environmentally induced neoplasm? |
X-irradiation. NO CURRENT LITERATURE SUPPORTING A ROLE OF CELLS PHONES WITH BRAIN TUMORS
-Low-level XRT to scalp for tinia capitis •XRT to other brain tumors •Prophylactic CNS radiation to children with acute lymphocytic leukemia (ALL) Meningioma. It is most frequently multiple and anaplastic and develops 10 years after exposure. [Two other environmentally-induced neoplasms: sarcomas and gliomas] |
|
|
Clinical presentation of those with brain tumors
|
Subacute presentation of focal neurologic deficit
•Seizure •Focal or generalized •Presenting symptom in 1/3 of brain tumors •Important to consider neoplasm following new onset of focal seizure •Most likely with tumors that invade or compress cerebral cortex compared to subcortical tumors •Nonfocal symptoms •Headache (particularly early-morning) •Dementia •Personality change •Gait disorder |
|
|
What is the most common imaging test for diagnosis and which is the best imaging test for diagnosis of intracranial neoplasm?
|
MOST COMMON: CT (relatively sensitive but not specific, can miss structural lesions; low grade gliomas are nonenhancing)
BEST: MRI Histological diagnosis also used; stereotactic biopsy or open craniotomy and resection |
|
|
What is a gangliocytoma?
|
A tumor that arises from ganglion cells in the CNS. Sometimes equated with ganglioneuroma. In this context the glial nature of the tumor is de-emphasized. Sometimes implies that the tumor is ENTIRELY NEURONAL
Often associated with seizures |
|
|
What is a ganglioglioma?
|
Tumors with a mixture of glial elements (looking like a low-grade astrocytoma) and mature-appearing neurons. Most of these tumors are slow growing, but the glial component occasionally becomes frankly anapestic, and the disease then progresses rapidly.
Often present with seizures |
|
|
*What is an astrocytoma?
|
A type of brain tumor that arises from astrocytes. There are several different categories of astrocytic tumors, the most common being fibrillary and pilocytic astrocytomas. Fibrillary astrocytomas account for about 80% of adult primary brain tumors
They are most frequent in the fourth through sixth decade Usually found in the cerebral hemispheres Commonly present with seizures, headaches, and focal neurologic deficits related to anatomic site of involvement They are graded based on the AMEN criteria: -Pilocytic astrocytoma (grade I) -"Low-Grade" astrocytoma (grade II) -Anaplastic astrocytoma (grade III) -Glioblastoma (multiforme) (grade IV) |
|
|
*Describe the onset of symptoms, age at diagnosis, MRI contrast enhancement, histological pathology, Ki-67 index (showing cells dividing), treatment options, and survival prognosis for low grade astrocytoma
|
Symptoms will take years to develop
Age of diagnosis is typically between 5-30 (younger) Can never see these tumors on MRIs Histological studies will only show nuclear atypia Only 2% of cells will be replicating/dividing Surgery or just observation (slow growing) are treatment procedures The prognosis averages around 5-10 years after onset |
|
|
*Describe the onset of symptoms, age at diagnosis, MRI contrast enhancement, pathology, Ki-67 index (showing cells not dividing), treatment options, and survival prognosis for anaplastic astrocytoma
|
Symptoms will take months to develop
Age of diagnosis is typically between 30-50 Can often be seen on MRIs Histological studies will show nuclear atypia and mitoses Only 5-10% of cells will be replicating/dividing Surgery and/or radiation are treatment procedures The prognosis averages around 3-4 years after onset |
|
|
*Describe the onset of symptoms, age at diagnosis, MRI contrast enhancement, pathology, Ki-67 index (showing cells not dividing), treatment options, and survival prognosis for glioblastoma
|
Symptoms will take weeks to develop
Age of diagnosis is typically >50 (oldest, elderly) Can always see these tumors on MRIs Histological studies will show nuclear atypia, mitoses, and endothelial proliferation/necrosis >10% of cells will be replicating/dividing Surgery, radiation, and chemotherapy are treatment procedures The prognosis averages around 1 year after onset |
|
|
*What is a Pilocytic Astrocytoma
|
Relatively benign tumors, often cystic, that typically occur in children and young adults and are usually located in the cerebellum. THEY ARE GRADE 1 IN THE AMEN CRITERIA
Presents with symptoms of hydrocephalus or mass effect Cured by surgical resection without adjuvant therapy in most cases Normal neurologic outcome and development common Pathology: "Piloid" glial cells with Rosenthal fibers |
|
|
*What is a Glioglastoma Multiforme (GBM)
|
Most common primary brain tumor and most aggressive of the gliomas
It is the least likely to respond to therapy Median age of onset is 62 years old (more common in elderly) Can either present as a secondary GBM or a primary/de novo GBM -Secondary GBM originated from a lower grade astrocytoma. It often presents after many years of seizures. Most likely occurs from well timed p53 hits. Presents in younger patients -Primary GBM: Symptoms develop over weeks to months. Seen more often in older patients. Typically occurs from EGFR amplification rather than p53 hits |
|
|
Describe Secondary glioblastoma multiforme. What makes it unique from the primary form?
|
Originates from a lower grade astrocytoma. It often presents after many years of seizures. Most likely occurs from well timed p53 hits. Presents in younger patients
|
|
|
Describe Primary glioblastoma multiforme. What makes it unique from the secondary form
|
Symptoms develop over weeks to months. Seen more often in older patients. Typically occurs from EGFR amplification rather than p53 hits
|
|
|
*What are the treatment options for glioblastoma multiforme?
|
Pretty Dismal
-Maximal surgical resection (Mass infilltration, really hard to get it all out) -Radiation -Chemotherapy (temozolomide)- better response if enzyme MGMT is Methylated and thus inactivated. Need to switch to a different therapy if it is unmethylated -Trials also out trying to create vaccine against the patients own antibodies |
|
|
*Describe Oligodendrogliomas
|
Tumors that originate from oligodendrocytes.
They constitute about 5% to 15% of gliomas and are most common in the fourth and fifth decades. They have several years of neurological complaints, often including seizures Lesions are found mostly in the cerebral hemispheres, with a predilection for white matter HAVE A BETTER PROGNOSIS THAN DO PATIENTS WITH ASTROCYTOMAS OFTER LOOK LIKE "FRIED-EGG CELLS" WITH "CHICKEN WIRE" VASCULATURE; OFTEN CALCIFIED -scant cytoplasm surrounded by a halo They are divided into "low-grade" (grade II) and anaplastic (grade III) tumors. Is no distinction between grade III and grade IV because they have similar prognosis |
|
|
*Describe a low-grade oligodendroglioma
|
Often radiologically indistinguishable from astrocytomas more likely to be CALCIFIED
• SEIZURE is common presentation • Survival—better than astrocytomas on average • Earlier studies--> 10 year median survival • More recent series--> 16 year median survival • Lead-time bias--> earlier diagnosis by MRI • Patients can often be followed for years without intervention STILL NOT A GOOD DISEASE, if you get it at 30 not very good |
|
|
Describe the prognosis for anaplastic oligodendroglioma and how it is treated
|
Prognosis better than for anaplastic astrocytoma
• Oligodendrogliomas are more chemosensitive • Chemotherapy regimens -PCV regimen (old) -Procarbazine -CCNU (lomustine) -Vincristine -Temozolomide (better response if enzyme MGMT is Methylated and thus inactivated) Tumors with deletions of chromosomes 1p and 19q typically respond to chemotherapy and have a better prognosis Only 25% of those lacking these markers respond Prognosis even better if MGMT is methylated, indicative of a better response to temozolamide |
|
|
How do the genetics work for treatment options for oligodendroglioma
|
Tumors with deletions of chromosomes 1p and 19q typically respond to chemotherapy and have a better prognosis
Only 25% of those lacking these markers respond Prognosis even better if MGMT is methylated, indicative of a better response to temozolamide |
|
|
*Describe Ependymomas
|
Tumors that arise from ependymal cells. Only defined by two grades: "Low-grade" (grade II) and anaplastic (grade III) ependymomas.
Tend to occur more often in younger patients, particularly the anaplastic variant Most often arise next to the ependymal-lined ventricular system, including the central canal of the spinal cord, most commonly found in the 4th venticle Constitutes 5% to 10% of the primary brain tumors in young adults In adults the spinal cord is their most common location, particularly frequent in the setting of neurofibromatosis type 2 In histological stains seen as perivascualr pseudorosettes (around blood vessel, see processes into the vessel) PROGNOSIS IS COMPLETELY DEPENDENT UPON THE EXTENT OF SURGICAL RESECTION (more you get the better the prognosis) Can radiate residual tumor IS CHEMORESISTANT (need to be aggressive to get out in front as much as possible |
|
|
*Describe Meningiomas
|
Second most common primary brain tumor.
◦ Origin--> meningothelial cells that form external membranous covering of brain ◦ Do NOT arise from brain parenchyma Classified as brain tumors ◦ Arise in intracranial cavity ◦ Present with neurologic symptoms Many meningiomas are asymptomatic and diagnosed at autopsy. Predominantly benign Most frequent radiation-induced intracranial neoplasm THEY USUALLY COME TO ATTENTION BECAUSE OF VAGUE NONLOCALIZING SYMPTOMS, OR WITH FOCAL FINDINGS REFERABLE TO COMPRESSION OF UNDERLYING BRAIN Variable symptoms that depend on where it arises from |
|
|
*What are the possible locations of meningiomas and how do they present
|
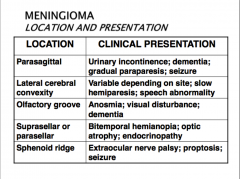
THEY USUALLY COME TO ATTENTION BECAUSE OF VAGUE NONLOCALIZING SYMPTOMS, OR WITH FOCAL FINDINGS REFERABLE TO COMPRESSION OF UNDERLYING BRAIN
Variable symptoms that depend on where it arises from |
|
|
Describe the histological presentation (pathology of meningiomas)
|
Unlike gliomas, vast majority are typical (grade I) and low-grade but often large when detected (slow growing, so large that they can invade soft tissues)
Atypical meningiomas (grade II) ◦ Increased cellularity and mitotic figures ◦ Brain invasion (need to invade to get to grade II) Anaplastic meningiomas (grade III) ◦ Sarcoma-like ◦ Many mitoses and necrosis Typical pathology ◦ Spindle cells concentrically arranged in a whorled pattern ◦ Psammoma bodies (laminated calficiations) |
|
|
*What are Psammoma bodies
|
They are laminated calcifications that when found with spindle cells concentrically arranged in a whorled pattern show that there is a meningioama
|
|
|
What is the treatment for Meningiomas?
|
Typical meningioma--> total surgical resection is often curative
◦ Recurrence risk After GTR--> <20% after 10 years Subtotal resection--> 60% at 5 years Radiosurgery--> if subtotal resection Atypical meningioma or anaplastic meningioma--> XRT or radiosurgery even if GTR achieved Chemotherapy after recurrence--> little success THE OVERALL PROGNOSIS OF MENINGIOMAS IS INFLUENCED BY THE SIZE AND LOCATION OF THE LISON< SURIGCAL ACCESSIBILITY, AND HISTOLOGICAL GRADE |
|
|
*Describe Schwannomas
|
-Third most common primary brain tumor
-A benign primary intracranial tumor of the myelin-froming cells of CN VIII -CNS: most often in vestibular division of 8th cranial nerve; misnomer “ACOUSTIC NEUROMA” -Schwann cell origin (cranial/peripheral nerve) -Bilateral 8th nerve schwannomas—diagnostic of neurofibromatosis type 2 -Surgical resection is curative (really long procedure, can take up to 12 hrs, try to preserve hearing) -Pathology: spindle-shaped cells in both loosely- and densely-cellular foci; focal palisading of nuclei |
|
|
*Describe Pituitary Adenomas
|
-Suprasellar tumor
-Two types: -NON SECRETOR: Bitemporal hemianopia from compression of the optic chiasm (macroadenoma), can lead to visual field cut out -SECRETOR: Endocrine abnormalities (microadenoma). Might be small but hormonally active, leads to endocrine malfunctioning -Prolactinoma is most common secretor -Treatment ◦ Surgery (intracranial and/or transsphenoidal) ◦ Dopamine agonists (prolactinomas), shrinks tumor ◦ Somatastatin analogs (growth hormone adenomas) ONCE YOU COME OFF THE THERAPY, THE TUMOR BALLOONS UP |
|
|
*Describe Medulloblastomas
|
-Malignant 4th ventricular tumor, seeds CSF pathways
-Occurs predominantly in children and exclusively in the cerebellum. Accounts for 20% of pediatric brain tumors -Normal and high risk groups (age, spread, degree of resection, pathology) -Always requires surgery + craniospinal XRT and chemotherapy. Highly malignant and prognosis is dismal without treatment -Today - 5 yr. survival in normal risk group > 75- 80% -Pathology: “small blue-cell tumor,” Homer Wright rosettes |
|
|
*Describe Craniopharyngioma
|
-A type of brain tumor derived from pituitary gland embryonic tissue
-Common suprasellar tumors of children and adults (bimodal distribution) -Locally invasive, difficult to treat, unpredictable response to multiple treatment types, treatment morbidity high (LOCATION). Processes go all over the place, makes it pathologically benign and histologically malignant -Origin: Rathke’s cleft/pouch, same place as pituitary adenomas, near hypothalamus and pituitary (NASTY LOCATION) -Pathology: stellate, “squamoid”-type epithelium; often calcified; cysts with “motor oil” fluid (cholesterol-rich) |
|
|
*Describe Hemangioglabstomas
|
-Tumors of the central nervous system that originate from the vascular system usually during middle-age
-Seen associated with Von Hippel-Lindau syndrome -Most often cerebellar -Can look identical to pilocytic astrocytoma radiologically, thus need to look under microscope -Can produce EPO--> secondary polycythemia -If resectable, surgery is curative -Pathology: lipidized, “foamy” cells with extensive vascularization. Very vascular and very bloody |
|
|
Where are pediatric brain tumors typically found?
|
Posterior fossa
- Cerebellar (usually pilocytic) astrocytoma - Brain stem astrocytoma (“pontine glioma”) - Medulloblastoma - Ependymoma Supratentorial - Astrocytoma, germ cell tumor, ganglioglioma, choroid plexus papilloma, oligodendroglioma, craniopharyngioma |
|
|
What is the relationship between pilocytic astrocytoma and childhood?
|
-Most common primary brain tumor in children
-Presents with symptoms of hydrocephalus or mass effect -Cured by surgical resection without adjuvant therapy in most cases -Normal neurologic outcome and development common -Pathology: “piloid” glial cells with Rosenthal fibers |
|
|
How common are brain metastases compared to other cancers in the brain? Why is there a rising incidence of these tumors?
|
Most common intracranial tumor
◦Considerably more than half of all brain tumors in adults ◦Develop in 10-30% of adults and 6-10% of children with cancer ◦Annual incidence 98,000-170,000 cases in US -10-15% of cancer deaths, indicates poor prognosis, survival <6 months -symptoms: headache, weakness, behavior changes, seizures Rising incidence due to: ◦Prolonged survival due to better systemic management ◦Ability of MRI to detect small brain metastases |
|
|
What are the most common cancers to met to the brain?
|
-lung (50%)
-breast (15-20%) -unknown (10%) -melanoma (10%) -colorectal cancer (5%) almost never: prostate, esophagus, oropharynx, cervix |
|
|
How to diagnose brain metastasis from unknown primary sites,
|
Complete physical examination with particular attention to
◦Skin for melanoma ◦Breast for masses ◦Rectal examination and fecal occult blood testing ◦Testicle CT of the chest, abdomen, and pelvis Blood tests ◦CEA ◦Liver function tests ◦AFP and β-hCG in young people (germ cell tumors) 30% of patients no primary can be identified, biopsy or resection for histologic diagnosis needed |
|
|
What are some patterns of brain metastases?
|
Parenchymal brain metastases 2/3
◦Most common multiple enhancing lesions of variable size associated with mass effect from vasogenic edema ◦Occasional solitary brain mass ◦Predilection to develop in cerebral gray-white matter junction and cerebellum Craniospinal “carcinomatous meningitis” ◦Adenocarcinomas lung or breast ◦Melanoma ◦High-grade lymphoma |
|
|
How do you treat brain metastases?
|
Multiple brain metastases palliative intent
◦Fractionated whole brain XRT ◦Gamma knife or stereotactic radiosurgery if <6 lesions all of which are <3.5 cm in diameter Solitary brain metastasis ◦Surgical resection Objectives Improve survival , reduce sxs of mass effect Possible cure ◦Gamma knife, if no mass effect symptoms |
|
|
What are epidural spinal metastases?
|
Spinal cord compression from metastases three patterns of spread:
◦Epidural spread of hematogenous vertebral metastases: vast majority of cases ◦Extension from paraspinal space through intervertebral foramina Lymphoma Renal cell carcinoma ◦Hematogenous dissemination: acute leukemia |
|
|
What are symptoms of spinal cord compression?
|
Back pain or radicular pain almost always initial symptom
Motor weakness present in about 75% of patients at diagnosis. Sensory loss/”level” approximately 50% at diagnosis Autonomic dysfunction ◦Urinary urgency and incontinence ◦Fecal incontinence ◦Impotence |
|
|
How do you treat spinal cord metastases?
|
Aggressive surgical resection
•Remove as much tumor as possible •Immediate decompression •Stabilize spine •Radiation •Combo does not significantly lengthen survival but does improve quality of life |