Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
22 Cards in this Set
- Front
- Back
|
Mendelian inheritance of Wilson’s(hepatolenticular degeneration) disease?
|
Autosomal recessive disorder
|
|
|
Pathogenesis of Wilson’s (hepatolenticular degeneration)disease ?
|
Pathogenesis
a. Gene mutation results in: (1) Defective hepatocyte transport of copper into bile for excretion (2) Decreased synthesis of ceruloplasmin (binding protein for copper in blood) b. Unbound copper eventually accumulates in blood (1) Loosely attached to albumin (2) Copper deposits in other tissues causing a toxic effect. |
|
|
Normal functions of ceruloplasmin and effect when decreased in Wilson’s (hepatolenticular degeneration) disease
|
Ceruloplasmin, the binding protein for copper, is secreted into the plasma where it represents 90-95% of the total serum copper concentration. The remaining 5-10% of copper is free copper that is loosely bound to albumin. Ceruloplasmin is eventually taken up and degraded by the liver. The copper that was bound to ceruloplasmin is excreted into the bile. The gene defect in Wilson's disease affects a copper transport system that produces a dual defect decreased synthesis of ceruloplasmin in the liver and decreased excretion of copper into bile. Accumulation of copper in the liver increases the formation of free radicals causing damage to hepatocytes. In a few years, unbound copper is released from the liver into the circulation (increased in blood and urine) where it can cause hemolysis or damage to the brain, kidneys, cornea, and other tissues.
|
|
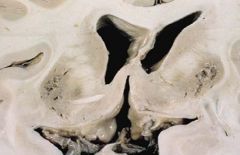
Gross findings (be able to recognize)of Wilson’s (hepatolenticular degeneration)disease:
|
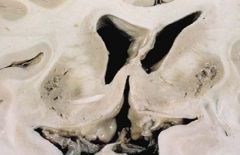
-Liver disease progresses from acute hepatitis to cirrhosis
-Kayser-Fleischer ring -Central nervous system disease |
|
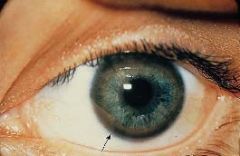
Clinical findings (be able to identify Kayser-Fleischer ring and exact site of copper deposition)of Wilson’s (hepatolenticular degeneration) disease.
|
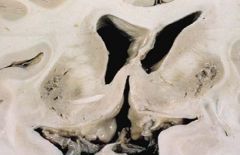
Kayser-Fleischer ring
•Due to free copper deposits in Descemet's membrane in the cornea Central nervous system disease (1) Atrophy and cavitation of basal ganglia, particularly the putamen (2) Copper deposits in the putamen • Produces a movement disorder resembling parkinsonism (3) Copper deposits in the subthalamic nucleus • Produces hemiballismus (4) Copper is toxic to neurons in the cerebral cortex • Produces dementia |
|
|
Laboratory findings of Wilson’s (hepatolenticular degeneration) disease:
|
Decreased total serum copper
• Due to decreased ceruloplasmin e. Decreased serum ceruloplasmin • Useful in diagnosing Wilson's disease in early stages of the disease f. Increased serum and urine free copper • Useful in diagnosing Wilson's disease in later stages of the disease |
|
|
Wilson's (hepatolenticular degeneration) disease:
|
↓ synthesis ceruloplasmin, ↓ excretion of copper in bile
|
|
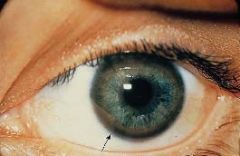
Kayser-Fleischer ring:
|
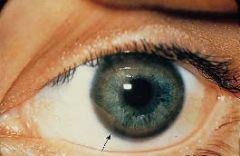
excess copper in Descemet's membrane of cornea
|
|
|
Mendelian inheritance of acute intermittent porphyria
|
Autosomal dominant disorder
|
|
|
enzyme deficiency and accumulation products in blood and urine in acute intermittent porphyria
|
a. Deficiency of uroporphyrinogen synthase (alias porphobilinogen deaminase)
b. Proximal increase in porphobilinogen (PBG) and δ-aminolevulinic acid (ALA) |
|
|
“window-sill test” for acute intermittent porphyria
|
Urine is colorless when first voided.
(1) Exposure to light causes oxidation of PBG to porphobilin producing port-wine color. (2) Classic "window-sill test" |
|
|
Effect of alcohol on heme in an individual with acute intermittent porphyria
|
Decreasing heme precipitates porphyric attacks by increasing porphyrin synthesis.
• Example: drugs enhancing liver cytochrome P-450 system (e.g., alcohol) |
|
|
clinical findings in acute intermittent porphyria
|
Neurologic dysfunction
(1)Recurrent bouts of severe abdominal pain simulating acute abdomen (2) Often mistaken for a surgical abdomen • Patient has “bellyful of scars”. b. Psychosis, peripheral neuropathy, dementia |
|

pathogenesis of acute intermittent porphyria
|

Defect in porphyrin metabolism
a. Deficiency of uroporphyrinogen synthase (alias porphobilinogen deaminase) b. Proximal increase in porphobilinogen (PBG) and δ-aminolevulinic acid (ALA) c. Urine is colorless when first voided. |
|
|
Acute intermittent porphyria:
|
“bellyful of scars”
|
|
|
what type of anemia is produced in a vitamin B12 deficiency?
|
macrocytic anemia (notes)
megaloblastic anemia (Robbins) |
|

sites of degeneration vitamin B12 deficiency
|

Subacute combined degeneration of the spinal cord:
-Dorsal column, -lateral corticospinal tract, -spinocerebellar tract demyelination |
|
|
clinical effects of a vitamin B12 deficiency
|
Dementia, peripheral neuropathy
|
|
|
Why administering and IV with glucose can precipitate an acute Wernicke’s syndrome and how it can be prevented.
|
It may precipitate an acute thymine deficiency in alcoholics (depleted by the pyruvate dehydrognase reaction). Deficiency is manifested by Wernicke-Korsakoff syndrome (RR Biochemistry p.55)
|
|
|
Pathogenesis of
Wernicke-Korsakoff syndrome |
Most often due to thiamine deficiency.
|
|
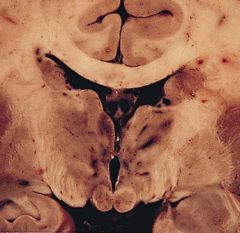
gross and microscopic findings (be able to identify)
Wernicke-Korsakoff syndrome |
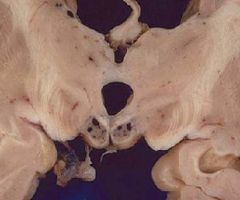
(1)Hemorrhages with hemosiderin deposits; sites
• Mamillary bodies, wall of the third and fourth ventricles (2)Neuronal loss, gliosis, vessel hemorrhage |
|
|
clinical findings in both parts of the Wernicke-Korsakoff syndrome
|
Wernicke’s encephalopathy:
reversible findings include- • Confusion, ataxia, nystagmus, ophthalmoplegia (eye muscle weakness) Korsakoff’s psychosis (1) Advanced irreversible stage of Wernicke’s encephalopathy •Targets the limbic system (2)Anterograde amnesia (inability to form new memories) (3)Retrograde amnesia (inability to recall old memories) |