![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
120 Cards in this Set
- Front
- Back
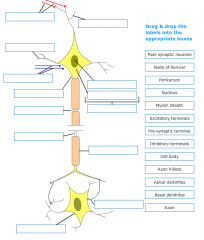
Label this diagram: |
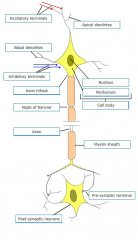
|
|
|
Where does protein synthesis occur in the neuron? |
The cell body consisting of a perikaryon and a nucleus |
|
|
Where do inhibitory inputs synapse on? What does this inhibitory input generate? |
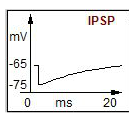
Graded hyperpolarisation, an IPSP |
|
|
What is the major role of the apical dendrites? |
Receive excitatory input from other nerve cells via axodendritic synapses. Activation of excitatory input generates a graded depolarisation - EPSP. N.B. summation of EPSPs at axonal hillock |
|
|
What occurs upon excitation of dendrites? |
Achieved via transmitter released from excitatory terminals - generates dendritic potential which may propagate passively or regeneratively to affect the excitability of the cell body.
If dendritic AP is generated it may trigger release from dendrites to affect excitability of neighbouring dendrites and/or presynaptic fibre terminals |
|
|
What are the ion concentrations in the axoplasm and the interstitial fluid? |

|
|
|
What does the resting nerve cell membrane have a high relative potential to? |
K+ = 1.0 CL = 0.3
Na = 0.01 |
|
|
What is the typical resting membrane potential of a central neurone? |
-65mV - resting membrane potential approximates K+ equilibrium potential - if K+ conc changes RMP changes |
|
|
What is the negative resting potential of the central neurone due to? |
K+ leaking out through non gated K+ channels.
K⁺ diffuses out of the cell down its concentration gradient. At equilibrium, outward K⁺ movement is balanced by the electrical gradient and no net K⁺ movement occurs. The electrical gradient when K⁺ is in equilibrium approximates to the Resting Membrane Potential (RMP). Na⁺ and Cl⁻ have little effect on the RMP. |
|
|
What is the equilibrium potential for Na+? |
The equilibrium potential for Na+ (ENa⁺) in a nerve cell is approximately +58 mV.
At equilibrium, the concentration gradient moves Na⁺ into the cell, which depolarizes the membrane. A membrane potential of +58 mV exactly opposes the concentration gradient and there is no net movement of Na⁺.
During the depolarization phase of the action potential the membrane potential tends towards ENa+ as the membrane permeability to ENa⁺ is greatly increased. |
|
|
Which neurotransmitters are peptides/non-peptides? |
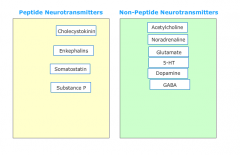
|
|
|
What categories do non-amine neurotransmitters fall into? |
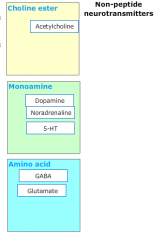
|
|
|
How are peptide NTs synthesised? |
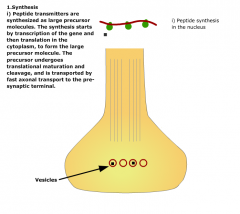
|
|
|
What are some features of non-peptide neurotransmitters? |
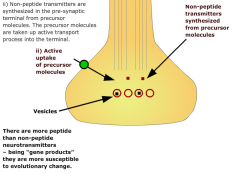
|
|
|
Where are peptide/non-peptide NTs stored? |
Sequestered in storage vesicles following entry by active transport. |
|
|
What happens upon terminal invasion via propagated action potentials? |
Depolarises terminal membrane This depol opens voltage gated Ca2+ channels allowing Ca2+ to enter the terminal down the EC gradient. |
|
|
When is cotransmission said to occur? |
When both peptides and nonpeptide NTs are simultaneously released |
|
|
How can NTs regulate their own release? |
Combine with pre-synaptic autoreceptors. |
|
|
How is non-peptide NT release terminated? |
By active reuptake into presynaptic terminal - when reuptaken can be degraded via enzymes or taken back up into storage vesicles. Can also be terminated by metabolic degradation or diffusion away from the synapse. |
|
|
What are retrograde transmitters? |
Some lipophilic modulators such as endocannabinoids and soluble gases e.g. NO, which use unconventional methods of transmission
Not stored in vesicles but synthesised on demand, act as retrograde messengers; released from post-synaptic site and acting on the pre-synaptic terminal |
|
|
What is the typical synaptic delay (i.e. time between depolarization of the pre-synaptic terminal and the onset of the post-synaptic response)? |
0.5 msec - average value and includes time taken for pre - synaptic Ca⁺⁺ channels to open, Ca⁺⁺ to trigger fusion of storage vesicles with the active zones of the terminal, transmitter release and diffusion across the synapse, interaction with post-synaptic receptors and initiation of the post-synaptic response. Some of these processes are extremely fast – channel opening usually takes less than 10 µseconds and diffusion of transmitters across the synapse less than 50 µsec |
|
|
What effect will a decrease in extracellular [Ca⁺⁺] have on synaptic transmission? |
Inhibit - The Ca⁺⁺ dependence of transmitter release, and hence synaptic transmission, is well-known. Mg⁺⁺, which competes with Ca⁺⁺ at the voltage gated Ca⁺⁺ channel, also inhibits transmission at sufficiently high concentrations (>5nM). There is a wide range of organic blockers of voltage gated Ca⁺⁺ channels, some of which are in clinical use. |
|
|
Approximately how many vesicles are there in each terminal? |
10000 This is the approximate number of vesicles in a single motor nerve terminal at the neuromuscular junction. Approximately 0.5 – 1% are utilised for each action potential that invades the motor nerve terminal. The number of vesicles per terminal in the central nervous system is more variable, but 10,000 is still a good approximation. |
|
|
How rapidly are peptide transmitters transported from the nucleus to the nerve terminal
|
400mm/day There are two systems which transport proteins from the nucleus to the nerve terminals; a slow system which transports cytoskeletal proteins and free cytosolic proteins (maximum rate of 2.5 mm/day) and a fast system which transports subcellular organelles, including peptide transmitters packaged into granules (maximum rate approximately 400 mm/day). |
|
|
What effect will blockade of the pre-synaptic autoreceptors have on synaptic transmission? |
Inhibitory and/or excitatory pre-synaptic autoreceptors may be present on nerve terminals, mediating a decrease or an increase in transmitter release, respectively, when activated by released transmitter. Because of this, we cannot predict the effect of blocking pre-synaptic autoreceptors on synaptic transmission |
|
|
Approximately how many molecules of transmitter are stored in each vesicle? |
3000 This is the upper limit of the number of molecules of acetylcholine stored in each vesicle in motor nerve terminals at the neuromuscular junction, the lower limit being approximately 1,000. Approximately 5,000 molecules of acetylcholine are needed to produce a miniature endplate potential and between 350,000 and 700,000 to produce an endplate potential. A single action potential invading the motor nerve terminal releases 150 quanta (i.e. the content of 150 vesicles) = between 150,000 and 450,000 molecules of acetylcholine. |
|
|
What effect will an inhibitor of transmitter inactivation have on synaptic transmission? |
Potentiate Transmitters are inactivated either by re-uptake into the nerve terminal, or metabolic degradation in enzyme catalyzed reactions, and sometimes by a combination of both. Such inactivation processes significantly reduce the half-lives of transmitters and hence limit their duration of action. Inhibitors of transmitter inactivation will therefore potentiate synaptic transmission; a number of such drugs are in clinical usage |
|
|
n the central nervous system, approximately how many vesicles fuse with the pre-synaptic nerve terminal in response to the influx of Ca⁺⁺ associated with each action potential? |
1-10 This is surprising, for the corresponding number at the neuromuscular junction is approximately 150. The reason for this is that each motor nerve terminal is large, with up to 300 active zones (sites where vesicles fuse with the pre-synaptic terminal to release their contents into the synaptic cleft). In contrast, nerve terminals in the central nervous system are much smaller, each typically possessing 1 or 2 active zones. |
|
|
What effect will blockade of the post-synaptic receptors have on the excitability of the post-synaptic neurone? |
Unpredictable The effect of blocking post-synaptic receptors at a given synapse will be unpredictable, unless it is known whether the transmitter is excitatory or inhibitory. |
|
|
What are ligand gated ion channels? |
Ligand gated ion channels are heterooligomeric proteins traversing the membrane, often consisting of four or five subunits. The time taken for the transmitter molecules to bind and the ion channel to open is short (<<0.1 msec). The duration of channel opening depends, in part, on the rate of dissociation of the transmitter molecules and the kinetics of channel gating. Amino acids induce fast neurotransmitter action in the CNS, but most monoamine and peptide neurotransmitters do not activate ligand-gated ion channels, and thus act more slowly, as neuromodulators. |
|
|
What is a GPCR? |
This is the second major type of neurotransmitter receptor. A G-(guanine nucleotide binding) protein couples the receptor to its effector (usually an ion channel or an enzyme). The receptor consists of a single polypeptide chain that traverses the membrane 7 times. When a single molecule of transmitter binds, the G protein becomes activated by the displacement of GDP by GTP, separates from the receptor and then dissociates into its constituent α-GTP and β and gamma subunits. |
|
|
Which part of the GPCR binds the GDP/GTP? |
a subunit |
|
|
What usually mediates synaptic excitation? |
The opening of channels permeable to Na+and K+ |
|
|
What is the threshold, EPSP, and peak membrane potential of a motor neurone? |
Threshold = -55mV EPSP = 0mV Peak = +40mV |
|
|
What else can membrane depolarisations result from? Other than EPSP. |
I) Influx of Ca⁺⁺ |
|
|
What is the difference between weak and strong stimuli on the afferent fibres? |
A weak stimulus activates a relatively small number of afferent fibres. The small, short duration depolarization is an excitatory post-synaptic potential (EPSP). EPSPs are localised, non propagated potentials which result from the transmitter released from the afferent fibres activating a population of ligand gated ion channels. Stronger stimuli recruit more fibres, releasing more transmitter, driving the membrane potential toward its reversal potential (approximately 0 mV). |
|
|
What are the function of inhibitory NTs? |
The opening of channels permeable to Cl⁻ and K⁺ usually mediates synaptic inhibition. Inhibitory transmitters usually combine with post-synaptic receptors to open channels permeable to Cl⁻ causing a hyperpolarization (Inhibitory Post-synaptic Potential, IPSP). |
|
|
What is buspirone? |
Buspirone, a selective agonist of one of the 5-HT receptor subtypes, is used in the treatment of anxiety. |
|
|
What are the effects of drugs that reduce GABA-mediated inhibitory transmission? |
Anxiogenic - drugs that provoke anxiety |
|
|
What are the effects of benzodiazepines? |
Potentiate effects of GABA at GABAa receptor complex |
|
|
How is 5HT synthesised? |
Tryptophan actively taken up into nerve terminal. Utilises large neutral amino acid carrier - subject to competitive inhibition from AA e.g. valine Tryptophan catalysed to 5HTP via tryptophan hydroxylase. 5HTP to 5HT via DOPA decarboxylase
|
|
|
How is presynaptic 5HT stored? |
Vesicles with specific proteins, vesicles recycled with new/recycled NT. A carrier molecule couples 5HT with Na uptake in ATPase dependent process Active uptake against conc gradient; involves carrier molecule within the vesicle gradient. Coupled to transport of Na and hydrolysis of ATP |
|
|
How is presynaptic 5HT released? |
Depolarisation of nerve terminals occurs when a propagated action potential invades the terminals. Influx of Na+ via voltage gated Na+ channels reverses the potential difference across the membrane - depolarisation - which opens population of voltage gated Ca2+ N type channels. Influx Ca2+ triggers quantal release via exocytosis fusion vesicles into active zones of terminal membrane |
|
|
How are 5HT receptors activated? |
Released 5HT diffuses acrosss synaptic cleft - binds to post synaptic 5HT receptors and presynaptic 5HT autoreceptors |
|
|
How is 5HT inactivated? |
Stored in vesicles - degraded by MAO and the product reduced to 5hydroxytrptophol. 5HT degraded by MAO and product oxidised to 5HIAA (5hydroxyindolacetic acid) or reduced to 5 hydroxytryptophol 5hydroxyindolacetaldehyde to 5HIAA via aldehyde dehydrogenase |
|
|
What are the 2 types of 5HT cell bodies and where are they found and where do they project? |
Found in raphe nuclei in pons and upper brainstem. 2 groups - caudal and rostral raphe nuclei
Caudal in lower BS - 5HT fibres project to medulla, cerebellum, dorsal horn of spinal cord
Rostral in upper BS - project to forebrain structures - hippocampus, BG, thalamus, limbic cortex, temporal lobe, CC |
|
|
What are features of 5HT function? |
The neurones have a characteristic pacemaker activity which is high during waking arousal and low during sleep. |
|
|
What is the nature of 5HT receptors? |
With the exception of the 5-HT3 receptor, which is a ligand-gated ion channel, all the other 5-HT receptors belong to the class of G-protein coupled receptors All five subtypes are negatively coupled to adenylate cyclase by a Gi/o protein. |
|
|
What are features of the 5HT1a receptor? |
High densites in limbic areas (regulation of mood and anxiety) and spinal cord (pain perception). Somatodendritic autoreceptors located on 5-HT neurones in the dorsal raphe nucleus inhibit 5-HT release. Post-synaptic receptors hyperpolarize neuronal membranes by direct activation of K+ conductances by Gi/o. |
|
|
What are features of the 5HT1b receptor? |
High densities in the striatum, hippocampus, cerebellum and layer IV of the cortex. There are inhibitory autoreceptors located on 5-HT neurones and inhibitory heteroreceptors located presynaptically on other neurones.
The drug sumatriptan binds to the 5-HT1DB (also known as the 5-HT1B) receptor |
|
|
What are features of the 5HT1d receptor? |
2 subtypes - 5-HT1Dα and 5-HT1Dβ. High densities in the olfactory system, striatum, cerebellum and parts on the limbic system. 5-HT1Dβ also known as 5-HT1B receptors are located on cranial blood vessels and are thought to be the target of the anti-migraine drug sumatriptan. |
|
|
What are features of the 5HT1E &F receptor? |
Both are found in high densities in the hippocampus and cortex. 5-HT1F receptors also found in other brain areas and in the uterus. |
|
|
What are general features of the 5HT2 receptor? |
All three subtypes are coupled to inositol phosphate turnover to increase formation of diacylglycerol and inositol-1,4,5-triphosphate. The 5-HT2A subtype is the classical 5-HT2 receptor. The 5-HT2C receptor corresponds to the receptor previously known as 5-HT1C. |
|
|
What are features of the 5HT2a receptor? |
Widespread distribution with high densities in the frontal cortex. Possible role in anxiety, depression and nociceptive transmission. In the periphery, it mediates contraction of vascular, tracheal and bronchial smooth muscle, platelet aggregation and increase in capillary permeability. Possible target for prophylaxis of migraine. |
|
|
What are features of the 5HT2b receptor? |
High levels in the cortex and some limbic structures and several peripheral tissues (gut, heart, kidney and lung). Mediates contractions of the rat fundic strip, a classical preparation for studying 5-HT, and some other GIT smooth muscle. Unknown role in the brain. |
|
|
What are features of the 5HT2c receptor? |
Widespread spinal and supra-spinal distribution. Potential roles include regulation of feeding, locomotion, hormone secretion, depression and anxiety. Also located on the endothelial cells of the choroid plexus where they probably influence the rate of formation of cerebrospinal fluid. |
|
|
What are features of the 5HT3 receptor family? |
Ligand gated, cation-selective ion channels which mediate membrane depolarization/neuronal excitation. Mediate inhibition of release of a number of CNS transmitters. High densities of post-synaptic receptors in the hippocampus, amygdala, area postrema, and some primary afferent terminals in the dorsal horn of the spinal cord. Mediate fast excitatory transmission. Peripheral location in the gastrointestinal tract. Roles in emesis and possibly anxiety, neuronal migration and differentiation. |
|
|
What are features of the 5HT4 receptor family? |
Positively coupled to adenylate cyclase by Gs. High levels in limbic structures and the striatum, but role in the CNS unknown. Mediate excitation in the CNS by reducing K+ and increasing Ca²+ conductances. In the periphery, they open voltage gated Ca²+ channels, directly relax oesophageal smooth muscle, indirectly contract other GIT smooth muscle (by releasing acetylcholine) and increase cardiac rate and force of contraction. |
|
|
What are features of the 5ht5 receptor family? |
These are shown in lower case because mRNA for these gene products exists but coupling to specific effector pathways has not been demonstrated.
Unknown transduction pathway, unknown roles. 5-ht5A receptors are found in the cortex, hippocampus and cerebellum. 5-ht5B receptors have a more limited distribution. Not expressed in the periphery.
|
|
|
What are general features of the 5HT6&7 receptors? |
Both receptors are positively coupled to adenylate cyclase (coupling to the Gs protein). No peripheral expression of the 5-HT6 receptor detected; low levels of peripheral expression of the 5-HT7 receptor. Several antidepressants bind with high affinity to the 5-HT6 receptor. Several neuroleptics bind with high affinity to the 5-HT7 receptor. |
|
|
What is the pharmaceutical use of L tryptophan? |
L-tryptophan used to be given either alone or as add-on therapy in the treatment of depression. The drug was withdrawn because of toxic effects (eosinophilia-myalgia syndrome). L-tryptophan is an essential amino acid which, because of its indole side chain, is highly hydrophobic. L-tryptophan is a constituent of proteins and in addition to being the precursor of 5-HT, can also be oxidised to kynurenine.
|
|
|
What are features of the tryptophan hydroxylase enzyme? |
Tryptophan hydroxylase requires molecular oxygen and reduced tetrahydrobiopterin. The enzyme is not saturated with tryptophan and so changes in substrate concentrations will affect the amount of 5-HT synthesized. The formation of 5-hydroxytryptophan (5-HTP) is rate limiting in the formation of 5-HT.
The drug p-chlorophenylalanine (pCPA) is a selective inhibitor of tryptophan hydroxylase and causes a reduction in the tissue levels of 5-HT; it has no clinical use. |
|
|
What is 5HT release modulated by? |
The release of 5-HT is modulated by inhibitory somatodendritic 5-HT1A receptors and inhibitory 5HT1B/D autoreceptors.
|
|
|
What drugs are agonists of the somatodendritic autoreceptors used to treat anxiety? |
Buspirone, ipsapirone and gepirone are relatively selective agonists of the somatodendritic autoreceptors. The drugs are used clinically to treat anxiety. Long-term treatment with buspirone desensitizes 5-HT1A somatodendritic autoreceptors, resulting in increased release of 5-HT and potentiation of 5-HT transmission |
|
|
What are characteristics of vesicular transporters? |
All are H+ pumping ATPases Drugs such as tetrabenazine compete with amines for their binding sites on amine transporters. Reserpine acts the same way - deplete levels of any amine whose vesicular storage is dependent on the same ATPase |
|
|
What is sumatriptan? |
Sumatriptan activates 5-HT receptors on the vascular smooth muscle (to induce vasoconstriction) and on the nerve endings (to reduce the release of neuropeptides). |
|
|
What is buspirone? |
Buspirone is a partial agonist of somatodendritic 5HT1A receptors and is used to treat anxiety. Buspirone, ipsapirone and gepirone are a family of structurally-related compounds. They all alleviate the symptoms of anxiety but the response to treatment may not appear for 2 weeks. This strongly suggests that their effects may involve a slow process of change in the receptors, which ultimately alters the dynamics of 5-HT transmission. |
|
|
What are 5HT receptor antagonists? |
Ondansetron is used in the management of nausea and vomiting induced by cytotoxic chemotherapy and radiotherapy and in post-operative nausea and vomiting. It is a selective 5-HT3 antagonist. Ondansetron, granisetron and tropisetron make up a family of structurally-related compounds. They have been reported to possess a range of behavioural effects but these are likely to reflect actions at other receptors. Ketanserin and ritanserin can act as antagonists at both 5-HT1A and 5-HT2 receptors. |
|
|
What is the effect of LSD on 5HT receptors? |
Experimental evidence now suggests that LSD selectively depresses the firing rate of raphe neurones; other evidence implies that its behavioural effects more closely correlate with the stimulation of the raphe systems. LSD itself is a non- selective agonist of 5-HT1 receptors.
|
|
|
What are features of the 5HT transporter? |
5-HT transporter The transporter is a single polypeptide chain that traverses the membrane 12 times. |
|
|
What are general features of tricyclics? |
Tricyclic drugs potentiate monoaminergic transmission. Most tricyclics inhibit amine re-uptake (e.g. amitriptyline, 5-HT; imipramine , 5-HT and noradrenaline (NA); desipramine , NA) . Some sensitize post-synaptic 5-HT receptors, others desensitize inhibitory alpha-2 adrenoceptors on 5-HT nerve terminals, and others potentiate NA transmission. |
|
|
What is RIMA? |
RIMA (e.g. moclobemide) differ from the irreversible MAOI, in that they are reversible and are selective inhibitors of MAOA. Because of this, they have a shorter duration of action and the “cheese reaction” is much less severe (the risk of such a reaction is around 10 times less than with an irreversible IMAO). |
|
|
What constitutes the limbic system? |
The limbic system of the brain is involved in emotional responses. It consists of a number of forebrain structures including:
Addiction depends on these structures |
|
|
What is the difference between physical and psychological dependence? |
There is no clear abstinence syndrome associated with psychological dependence.
Physical dependence is: |
|
|
What are examples of withdrawal effects from opiates, barbiturates, benzos, and alcohol? |
Opiates Barbiturates Benzodiazepines Alcohol |
|
|
What are the 2 main types of tolerance? |
1. Acute tolerance
Not all drugs to which tolerance develops cause dependence, e.g.: Tolerance develops to: However, these drugs do not cause dependence. |
|
|
Describe acute tolerance, relate this to the effect of taking nicotine. |
Acute tolerance or tachyphylaxis is a short lasting tolerance which occurs when a drug acts at a receptor which becomes desensitised by the first dose. Acute tolerance occurs with nicotine in tobacco. Nicotine causes depolarising block at some of the nicotinic receptors at which it acts. Over the smoking day, the nicotine- induced increase in heart rate (HR) diminishes. However, this effect recovers following overnight abstinence. |
|
|
What are the 2 categories of drugs on the brain? |
|
|
|
What are the features of nicotine? |
However, in comparison with amphetamine and cocaine, nicotine is a relatively mild psychostimulant. Nevertheless, nicotine and tobacco smoke have been demonstrated to: - increase alertness |
|
|
What are features of the dependence of nicotine? |
Therefore, the dependence liability of nicotine in tobacco is relatively high. During abstinence, the major withdrawal effects which may be experienced are: |
|
|
What 3 types of tolerance occur in nicotine use? |
1. Acute tolerance – many of the effects of nicotine, e.g. increase in heart rate and locomotor stimulation, show acute to short lasting tolerance to the drug due to desensitisation of the nicotinic receptors mediating these effects. |
|
|
How does nicotine act and what substance can block this effect? |
Nicotine stimulates nicotinic acetylcholine receptors in the brain to produce its rewarding effects, since mecamylamine, a nicotinic receptor antagonist which can enter the brain, blocks the psychomotor stimulant and pleasurable effects of smoking. |
|
|
How does nicotine affect the brain? |
Stimulates NAChR in VTA on dopaminergic neurones - release of dopamine in NA - this is how dependence develops.
Stimulation of NAChR in hippocampus increases attention and may underlie the improvement seen with nicotine.
Stimulation increases alertness in reticular formation |
|
|
What are the effects of benzodiazepines? |
Anxiolytics – benzodiazepines are powerful anxiolytic drugs in man and animals. They are also used to tame animals, to allow them to be handled more easily.
|
|
|
What benzos are there? |
Diazepam – is a long acting drug not only because it has a relatively long half-life (~32 h) but also because it has at least two metabolites which are also active i.e. N-desmethyldiazepam (nordiazepam)(T1/2=~60 h) and oxazepam (T1/2=~8 h). |
|
|
What is the nature of benzodiazepine dependence? |
Anxiety Iatrogenic |
|
|
What is the nature of benzodiazepine tolerance? |
Tolerance of the cellular type, does occur to a limited extent following long term treatment with the benzodiazepines. However, this does not seem to lead to patients increasing their dosage. In fact the anxiolytic action for which many people are prescribed benzodiazepines is not as susceptible to tolerance. The main clinical tolerance appears to develop to the anticonvulsant actions of the drugs. |
|
|
How do benzos act? |
Benzodiazepines (BDZs) bind to the GABAA receptor. Their binding causes an allosteric (structural) modification of the receptor that results in an increase in GABAA receptor activity. BDZs do not substitute as an agonist for GABA, but increase the frequency of channel opening events in the presence of GABA, which leads to an increase in chloride ion conductance and inhibition of the action potential. |
|
|
How do benzos & barbs have their function? |
Reticular formation - inhibit neurones to cause sedation
Raphe nuclei - stimulation of GABAa receptors in dorsal raphe nuclei attenuates the firing of 5HT neurones to mediate an anxiolytic effect |
|
|
How do barbiturates cause their effect? |
Thiopentane – an induction agent in general anaesthesia
Barbiturates will depress the activity of all cells such that at high doses, they depress the cardiovascular and respiratory systems causing death. However, their effects are more selective for cells in the CNS. Over the clinical dose range, the effects they produce vary from anxiolytic to general anaesthesia with increasing doses. |
|
|
What are the main effects of barbiturates? |
Euphoria – with most of the barbiturates taken orally, feelings of euphoria or well-being are experienced before sedation occurs |
|
|
What are features of barbiturate dependence? |
The chronic administration of clinically relevant doses of barbiturates leads to dependence in a large proportion of patients. Therefore, barbiturates have a high dependence liability. The dependence is both physical and psychological such that abrupt cessation of treatment produces a clear cut withdrawal syndrome, with the following symptoms occurring to a lesser or greater degree depending on the accustomed dose.
nxiety – this is one of the first symptoms to appear after withdrawal of barbiturates and for those who were prescribed barbiturates as anxiolytic or antidepressant drugs, this may be a rebound re-emergence of the condition for which they were treated. onfusion – this occurs before delirium tremens has fully developed. |
|
|
What are features of the 2 GABA receptors in the brain and the effect of barbiturates on them? |
GABAA receptor: |
|
|
What is the nature and stages of alcohol dependence? |
a small (~10%) proportion become truly dependent on it and develop alcoholism
Phase 1 Phase 2 Phase 3 |
|
|
Describe the 3 types of tolerance that occur with alcohol consumption. |
Acute tolerance – this is seen after one drinking bout, where the effects wear off even when the blood alcohol level is maintained.
Cellular tolerance – this occurs with regular drinking, where one has to consume more alcohol to produce the motor impairment seen in an abstainer. It occurs with or without dependence. - DUE TO CALCIUM CHANNEL UPREGULATION Acute alcohol, like many of the anaesthetic drugs, increases membrane lipid fluidity. This effect is reduced following repeated exposure to alcohol. Interestingly, alcoholics require larger doses of anaesthetics, suggesting cross tolerance occurs with this effect. However, the respiratory depressant effects of alcohol and other sedative drugs do not show the same degree of cross tolerance as seen with the anaesthetic effects. Pharmacokinetic tolerance – this only occurs in severe alcoholics. |
|
|
What is the order of reaction in alcohol metabolism? |
This is because alcohol dehydrogenase, the main enzyme for metabolising alcohol, has an apparent Km for alcohol of ~8mg/100ml of blood. Since a pint of beer will produce blood levels of ~35 mg/100 ml, then the rate limiting enzyme is saturated and the reaction progresses at a constant rate until the blood level falls to levels of ~8 mg/100 ml. |
|
|
How does alcohol have its effect in the brain? |
Selectively acts in the RF depressing firing of inhibitory ascending fibres - initial disinhibitory behaviour
Higher doses of alcohol depress the activity of cortical neurones - hypnotic effects + loss of consciousness |
|
|
How does cannabis act? |
The active ingredient of cannabis is delta-9-tetrahydrocannabinol (9-THC) and it is thought to exert its effect by binding to cannabinoid CB1 receptors in the brain. 9-THC binding to CB1 receptors activates G-proteins, that can ultimately activate or inhibit a number of signal transduction pathways. The G-proteins directly inhibit N and P/Q-type voltage dependent calcium channels and sodium channels, and indirectly inhibit other calcium channels via inhibition of adenylate cyclase. 9-THC binding and G-protein activation also can ultimately activate inwardly rectifying potassium channels and the MAP kinase signalling pathway. |
|
|
List some opiates and give their features? |
Morphine Opium is extracted from the poppy, Papaver somniferum, and has been used socially and medicinally for thousands of years. Morphine was first isolated in 1803 and used clinically as an analgesic and for the treatment of diarrhoea. It was freely available such that by the 1850’s there were – 20,000 shops which sold ~400 different opiate preparations. As laudanum, it was widely used by Britain’s poor since it was cheaper than alcohol. The dependence on opiates escalated and in the 1920’s controls were introduced in Britain on the importation, production and distribution of opiates. |
|
|
What are the overall effects of opiates? |
Analgesia – Opiates are very potent painkillers and this is the main clinical indication of use for these drugs especially post-operatively and in the relief of the pain of terminal cancer. |
|
|
How do opiates act? |
Opiates act on specific opioid receptors of which there are three main types:
|
|
|
What are endogenous opiate ligands? |
Beta – endorphin – This is a 31 amino acid peptide which is equally active at all three opioid receptors.
|
|
|
What are the dependence producing properties of opiates due to? |
inhibit nerve cell firing |
|
|
How do opiates act on the brain? |
NA - mu-opiate receptors mediating euphoria and dependence PAG - stimulation of mu-opiate receptors in this brain region contributes to analgesia. Spinal cord - kappa receptors directly cause analgesia VTA - mu-opiate receptors increase dopamine firing in NA - euphoria and dependence RF - all types of opiate receptors causes sedation and contributes to resp depression Area postrema - chemoreceptor trigger zone - stimulation of opiate receptors in brain causing N&V |
|
|
What are the effects of cocaine? |
Causes euphoria
Cocaine is a relatively potent local anaesthetic which is still used clinically as an ophthalmic anaesthetic, where it is applied locally, to the cornea, rendering it insensible. In addition, it causes dilatation of the pupils, mydriasis, and reduces intra-ocular pressure due to its potentiation of catecholamine activity. |
|
|
What features of dependence does cocaine have? |
The main symptoms of withdrawal are:
Dependence liability is the potential of the drug to induce dependence in people who self administer the drug chronically. Great dependence liability is seen in drugs which will induce dependence in a majority of users after relatively short periods of chronic treatment. For example, cocaine has great dependence liability since virtually all people who experiment with cocaine daily for a few weeks will become dependent on the drug. |
|
|
Describe tolerance of cocaine. |
The development of tolerance to cocaine is rather complex, with tolerance occurring to some but not all of the effects of cocaine. For example, many cocaine addicts will go on ‘cocaine binges´ to experience and try to maintain a ‘high’. However, the addict becomes tolerant to the euphoric effect and will take larger and larger doses to maintain their ‘high’. This type of excessive usage can lead to a cocaine- induce psychosis. This can result in violent behaviour due to paranoia, where the addict imagines they are being threatened or persecuted by innocent bystanders. Cocaine induced psychosis |
|
|
How does cocaine act? |
n common with amphetamine, the CNS stimulant actions of cocaine are due to its ability to potentiate the actions of the catecholamines in the brain. However, unlike amphetamines, it does not cause a direct stimulation of catecholamine release. It shares only one mechanism of action with amphetamines which is the following: Cocaine inhibits the uptake of dopamine (DA) but, unlike amphetamine, cocaine is not a substrate for the DA uptake transporter. |
|
|
What is the difference between cocaine and amphetamine administration when TTX, a sodium influx blocker, is given? |
Amphetamine stimulates dopamine release directly by a mechanism which is independent of nerve cell firing. Therefore, amphetamine will increase extracellular fluid (ECF) dopamine levels in the brain with or without TTX. In contrast, cocaine does not stimulate the release of dopamine directly. The increase in extracellular dopamine levels, normally seen in response to cocaine, is due entirely to inhibition of the uptake of dopamine which is released because the nerves are firing. Therefore, if nerve cell firing is inhibited by TTX, cocaine is ineffective. |
|
|
Where does cocaine act in the brain? |
NA - cocaine stimulates causing euphoria + plays a major part in dependence Hypothalamus - cocaine induced stimulation of nuclei in hypothalamus - increases temp + decreases food consumption RF - Stimulation - increases alertness |
|
|
Describe the nature of caffeine tolerance. |
In humans, cellular tolerance is reported to occur to some of the less desirable effects of caffeine after short periods of chronic use, e.g. people who do not normally drink coffee can experience palpitations, irritability and nervousness after consuming a strong cup of coffee. However, these effects appear to show tolerance within a matter of days of regular consumption. The psychostimulant effects of caffeine appear to show little tolerance and therefore people do not normally binge on coffee the way they binge on cocaine or amphetamine. However, there is no psychosis associated with excessive intake of caffeine which can occasionally occur, especially when students are burning the midnight oil before exams (!). |
|
|
How does caffeine work? |
Caffeine is a member of a class of compounds called methylxanthines which have the following primary sites of action: They inhibit phosphodiesterase (the enzyme hydrolyses cyclic AMP to the inactive 5’AMP). Both of these actions result in an increase in cyclic AMP, a second messenger for certain G-protein coupled receptors including some catecholamine receptors e.g. the beta-adrenergic and dopamine type 1 (D1) receptors. It is likely that the psychostimulant effects of caffeine are related to its ability to potentiate catecholamines in the brain due to potentiation of the cyclic AMP – dependent catecholamine receptors. |
|
|
What is the effect of caffeine on the different parts of the limbic system? |
NA - increases dop turnover, mediates pleasure of caffeine RF - Increases formation and arousal - not same as amphet/cocaine |
|
|
What are amphetamines? Examples? |
Amphetamine
They are powerful psychostimulants which: |
|
|
What is the nature of dependence of amphetamines? |
The amphetamines have powerful dependence liability such that chronic exposure very readily produces dependence. This occurs when the drug is administered orally or intravenously and manifests itself as psychological dependence but not physical dependence since there is no characteristic withdrawal syndrome evident on abrupt withdrawal. Dependence liability is the potential of a drug to induce dependence in people who self- administer the drugs chronically. Great dependence liability is seen with drugs which will induce dependence in a majority of users after relatively short periods of chromic treatment. For example, cocaine has great dependence liability since virtually all people who experiment with cocaine daily for a few weeks will become dependent on the drug. |
|
|
Describe the nature of amphetamine tolerance. |
Tolerance of the cellular type develops rapidly to many of the effects of the amphetamines.
Many amphetamine addicts inject amphetamine (commonly known as speed) intravenously in order to experience an intense euphoria of ‘flash’ and are known as ‘speed freaks’. With this type of excessive use, tolerance can develop to the euphoric effects of amphetamines, which leads the addict to keep escalating the dose to overcome the tolerance. This frequently leads to psychosis with symptoms similar to those seen in schizophrenia.
|
|
|
What are the effects of excessive stimulation and how long can they be maintained? |
The symptoms of psychosis are similar to those seen in schizophrenia, i.e.: Paranoid delusions Addicts can only maintain this type of excessive administration for days or a couple of weeks at most before 'crashing' into deep sleep and depression, probably due to exhaustion of the stores of neurotransmitters stimulated by amphetamines. |
|
|
How do amphetamines potentiate the effects of catecholamines? |
1. Stimulate release of catecholamines
Uptake Release MAO |
|
|
How do amphetamines affect different parts of the brain? |
Amphetamine induced stimulation of the NA - euphoria + major part in developing dependence
Hypothalamus - amphetamine induced stimulation of hypothalamic nuclei - increased temperature, decreased food consumption
RF - amphetamine induced stimulation of the RF - increased alertness |
|
|
Which drugs cause physical dependence? |
Alcohol, barbiturates, benzodiazopines, caffeine and opiates |