Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
85 Cards in this Set
- Front
- Back
|
What is amino acid metabolism?
|
Protein break down through sequential enzyme digestion
|
|
|
What is a protease?
|
Enzyme that catalyses the hydrolytic breakdown (hydrolysis - chemical breakdown due to reaction with water) of proteins into peptides or amino acids (proteolysis)
|
|
|
What is an endopeptidase?
|
An enzyme that breaks peptide bonds other than terminal ones in a peptide chain
|
|
|
What is an exopeptidase?
|
A peptidase that catalyses the removal of the last (carboxypeptidases) or first (aminopeptidases) amino acid from a peptide chain.
|
|
|
What enzyme does the stomach secrete?
|
Pepsin (an endopeptidase) in its inactive form (zymogen) pepsinogen
|
|
|
How is pepsinogen activated?
|
Cleavage (by HCl) of a peptide fragment from its amino terminus
|
|
|
When does this activation occur?
|
Autoactivated by active pepsin
Also, when pH of stomach lumen <5 - acidity denatures proteins so they are more susceptible to hydrolysis |
|
|
What are serine proteases(/endopeptidases)?
|
Enzymes that cleave peptide bonds in proteins in which serine is one of the amino acids in the enzyme's active site.
|
|
|
What enzymes does the pancreas secrete?
|
Serine endopeptidases - Trypsin, chymotrypsin and elastase.
Exopeptidases - carboxypeptidases A&B |
|
|
Where do these enzymes act?
|
Work in the lumen of the small intestine
Neutral conditions due to secretion of bicarbonate rich pancreatic juice |
|
|
In what form are all pancreatic proteases secreted in?
|
Inactive precursors - (trypsinogen, chymotrypsinogen, proelastase, procarboxypeptidase A&B)
|
|
|
How is trypsinogen activated?
|
By enteropeptidase (also called enterokinase)
By active trypsin (autocatalytic process) |
|
|
What releases enteropeptidase?
|
Secreted from enterocytes (the epithelial cells of the small intestine)
|
|
|
How are the other inactive precursors secreted by the pancreas activated?
|
Cleavage with trypsin
|
|
|
What is left after digestion by pancreatic enzymes?
|
Mix of amino acids and small peptides up to 6 amino acids long (oligopeptides)
|
|
|
Diseases which interfere with pancreatic secretion... (2) - associated problems
|
Pancreatitis and CFTR - prevent proper protein digestion resulting in protein malabsorption leading to malnutrition
|
|
|
What is the treatment for reduced pancreatic secretion?
|
Supply either extra exogenous pancreatic enzymes or dietary supplements of easily digested proteins
|
|
|
After pancreas where do digestive enzymes come from and at?
|
Brush-border membrane of enterocytes
Lumen of small intestine |
|
|
What enzymes does the brush-border contain?
|
Endopeptidases, aminopeptidases and dipeptidases
|
|
|
After brush-border enzyme digestion, what is left?
|
Mixture of dipeptides, tripeptides and amino acids
|
|
|
What takes up di- and tripeptides into enterocytes?
CLINICAL: What else is this transporter responsible for? |
Proton-coupled transporter
Absorption of beta-lactam antibiotics |
|
|
What absorbs amino acids into enterocytes?
|
A number of mainly sodium-coupled transport systems
|
|
|
What happens to di- and tripeptides once in enterocytes?
|
Cleaved by intracellular peptidases into free amino acids
|
|
|
What happens to amino acids in enterocytes?
|
Released via the basolateral membrane and enter the circulation
|
|
|
What are essential amino acids? Name them
|
Those that the body cannot make - alanine, aspartate, asparagine, cysteine, glutamate, glutamine, glycine, prolein, serine and tyrosine.
|
|
|
Uses of amino acids
|
Protein synthesis, hormone production e.g. adrenaline, neurotransmitter synthesis.
Deaminated - remaining carbon skeleton can then be either oxidised via TCA cycle, converted into glucose via gluconeogenesis or turned into fatty acids (not all are capable of all these fates) |
|
|
Which amino acids are glucogenic?
|
Those which can be degraded to pyruvate or TCA cycle intermediates
|
|
|
Which amino acids are ketogenic?
Name the two which are solely ketogenic |
Those which are convert into acteyl-CoA or acetoacetyl-CoA
Leucine and lysine |
|
|
What is the term for amino acids which are both gluco and ketogenic?
|
Mixed
|
|
|
Points of entry of amino-acid carbon skeletons into the citric acid cycle and into ketone body synthesis...
|
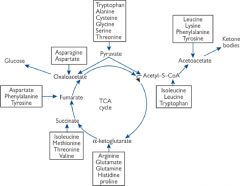
|
|
|
Where are excess amino acids stored?
|
They cannot be stored
|
|
|
First step of amino acid oxidation
|
Remove amino group (deamination) - excreted as urea
|
|
|
How is the amino group removed?
|
Transamination - catalysed by aminotransferases (transaminases)
Each amino acid has its own specific aminotransferase |
|
|
What is the overall reaction for transamination?
Two features of the reaction |
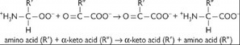
Amine group of an amino acid and keto group of a keto acid are exchanged
Easily reversible, requires no energy input |
|
|
What is the co-factor of all aminotransferases?
|
Pyridoxal phosphate (vitamin B6 derivative)
|
|
|
Most common amino group acceptor?
What does this form and how is the product used? |
Alpha-ketoglutarate
Glutamate - provides pool of amino groups for non-essential amino acid synthesis and deamination. |
|
|
Two other amino group acceptors and what they form as a result
|
Pyruvate --> alanine
Oxaloacetate --> aspartate |
|
|
What deaminates glutamate
|
Glutamate dehydrogenase
|
|
|
What is the benefit of pooling excess amino groups into glutamate?
|
Only one deamination pathway is required
|
|
|
Overall deamination reaction and where it takes place
|
Removal of amine group
Mitochondria of liver cells |
|
|
Main fate of ammonium (NH4+) from deamination reaction?
|
Incorporation into urea for excretion
|
|
|
What allosterically regulates glutamate dehydrogenase and why?
|
Increases in ADP and GDP - these compounds signal that amino acids need to be used as an energy source.
|
|
|
Other sites of NH4+ production (3)
|
Brain, muscle, intestinal cells
|
|
|
How are ammonium ions produced in the brain?
|
Inactivation (through breakdown) of neurotransmitter GABA into succinate and NH4+
|
|
|
How do ammonium ions produced in the brain enter the urea cycle?
|
One NH4+ combined with alpha-ketoglutarate to produce glutamate and then another incorporated to form glutamine --> transported to liver where it is deaminated and enters urea cycle
|
|
|
How are ammonium ions produced in muscle? (3)
|
Natural protein turnover
Muscle catabolism during starvation Breakdown of excess ADP during extreme exercise - 2ADP ----> ATP + IMP + NH4+ |
|
|
What happens to the NH4+ produced in muscle?
|
Combines with alpha-ketoglutarate to form glutamate - used to transaminate pyruvate, forming alanine and regenerating alpha-ketoglutarate.
|
|
|
What is the fate of alanine produced from the transamination of pyruvate in muscle cells?
|
Released into the bloodstream and taken up by the liver.
Deaminated, producing pyruvate which can either be oxidised in the TCA cycle or used for gluconeogenesis. |
|
|
What happens to the NH4+ produced in intestinal cells (enterocytes)?
|
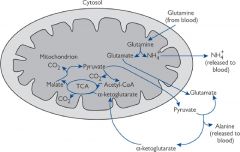
Same as brain, NH4+ combined with alpha-ketoglutarate to produce glutamate and then another incorporated to form glutamine - this serves as an energy source for the cell.
|
|
|
What are the two forms in which excess nitrogen can be excreted?
|
Urea or ammonium ions
|
|
|
Where in the body is urea generated and why?
|
In the liver as a soluble, non-toxic way of eliminating excess ammonia
|
|
|
What else can deaminate glutamate?
|
The renal cortex
|
|
|
How is ammonium used in the renal cortex?
|
Ammonium is used to assist with acidifying urine.
|
|
|
Why is the mechanism for deamination of glutamate in the renal cortex beneficial?
|
Mechanism conserves HCO3- which would otherwise need to be used in urea synthesis - would exacerbate any acidosis.
|
|
|
What happens if protein intake is greater than need?
|
Cannot store excess amino acids - carbon skeletons are used/stored and unwanted amino groups are excreted.
|
|
|
What happens if protein intake is less than need?
|
Protein catabolism to free carbon skeletons for energy - the amino groups produced need to be excreted
|
|
|
Why do blood ammonia levels need to be kept low?
What is the normal value? |
Toxic
(25-40 μM - micro molar) |
|
|
What happens if ammonium ion levels rise?
|
NH4+ reacts with alpha-ketoglutarate to form glutamate
|
|
|
What are the consequences for the brain of raised NH4+ levels?
|
Resulting raised glutamate levels reduce the rate at which ATP can be formed, starving brain cells of energy
|
|
|
What is 'nitrogen balance'
|
State of healthy adults - around 80% of excess nitrogen excreted as urea (remainder in free ammonium ions and creatine)
|
|
|
Where is most urea synthesised? (Organ)
Why is rate of synthesis strictly controlled? |
In the periportal cells (those surrounding the portal vein) or the liver.
To prevent ammonia build up |
|
|
The urea cycle
|
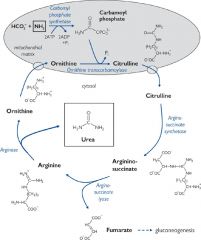
Formation of urea from one free ammonium ion and one donated from aspartate
|
|
|
Where does the urea cycle take place? (Cell)
|
Partly in the mitochondrial matrix, partly in the cytoplasm
|
|
|
What two amino acids does the urea cycle include that are not found in proteins?
|
Ornithine and citrulline
|
|
|
What are the two levels of urea cycle control?
|
Acute and chronic
|
|
|
What enzyme controls acute regulation of the urea cycle and what is this regulated by?
|
Carbamoyl-phosphate synthetase is regulated by the concentration of the allosteric activator N-acetyl-glutamate
|
|
|
What forms N-acetyl-glutamate and what stimulates this?
|
N-acetyl-glutamate synthase - activity stimulated by arginine (intermediate of the urea cycle)
|
|
|
What controls chronic regulation of the urea cycle? Time period?
|
Induction of urea cycle enzymes over 24-36 hours
|
|
|
What triggers chronic regulation?
|
Increased levels of ammonia in liver cells
|
|
|
What are the consequences for enzyme synthesis of prolonged or severe starvation?
|
Amino acids deaminated to use carbon back bone for energy - this may cause protein (and so enzyme synthesis) to be compromised
|
|
|
What is the treatment if control of the urea cycle is not functioning correctly?
|
Reduce protein level in diet and give a compound which aids nitrogen excretion (either through stimulating urea cycle or another compensatory pathway)
|
|
|
EXTRA What is the most common urea cycle disorder?
|
Ornithine transcarbamoylase deficiency
|
|
|
EXTRA Which gender is most severely affected by ornithine transcarbamoylase deficiency and why?
|
Generally males because the disease is X-linked
|
|
|
EXTRA What are the symptoms of ornithine transcarbamoylase deficiency and what do these cause?
|
Raised ammonia and amino acid levels, high blood or ororatic acid levels.
Mental retardation and can cause dead. |
|
|
EXTRA What is the treatment for ornithine transcarbamoylase deficiency and why?
|
Large quantities of benzoate and phenylacetate:
Benzoly-CoA reacts with glycine to form hippurate Phenylacetyl-CoA reacts with glutamine to form phenylacetylgluatime These products act as excretable substitutes for urea in the disposal of nitrogen. |
|
|
Which organ is the main site of deamination (amino acid degradation) and urea synthesis?
|
The liver
|
|
|
Which organ is the main site of gluconeogenesis during fasting? What fuels this process?
|
The liver
Carbon skeletons from amino acids |
|
|
What is the function of glutamate reductase?
|
Synthetic enzyme for citrulline
|
|
|
Which are the only cells to contain glutamate reductase?
|
Enterocytes
|
|
|
What is the fate of citrulline produced in the gut?
|
Metabolised to arginine in the liver - this is then converted to ornithine to increase the capacity of the urea cycle during times of increased protein intake.
|
|
|
What happens to muscle protein during fasting and starvation?
|
Broken down to provide carbon skeletons for gluconeogenesis in the liver
|
|
|
What are the main amino acids released from muscle protein breakdown?
|
Alanine and glutamine
|
|
|
What is the fate of alanine released from muscle protein catabolism?
|
Transported by the circulation to the liver for deamination and gluconeogenesis
|
|
|
What is the fate of glutamine released from muscle protein catabolism?
|
Transported to small intestine by circulation, taken up by enterocytes for energy and released as alanine
|
|
|
Glucose-alanine cycle
|
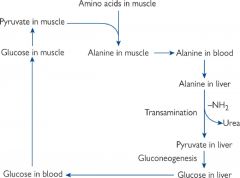
Transport nitrogen to liver as alanine and glucose back to muscles
|