Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
243 Cards in this Set
- Front
- Back
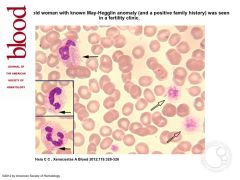
what is it
|
may-hegglin anomaly
very large, fairly well defined plae, basophilic, oval to round inclusions (3-6 um), 1-2 per cell |
|
|
what is the underlying defect in May Hegglin anomaly
|
nonmuscle myosin heavy chain A (MYH9) mutation
|
|
|
what is the inheritance pattern for May Hegglin anomaly
|
autosomal dominant
|
|
|
what hematologic findings coincide with May Hegglin anomaly
|
- thrombocytopenia;
giant platelets |
|
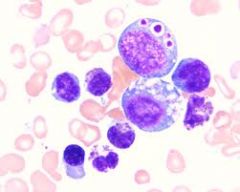
what is it
|
large, well defined round to irregular blue to green gray granules, 1-4 uM, approximately 10 inclusions per cell
|
|
|
underlying defect in chediak Higashi syndrome
|
abnormal fusion of lysosomal membranes
|
|
|
inheritance pattern for chediak higashi syndrome
|
autosomal recessive
|
|
|
other hematologic manifestations of chediak higashi syndrome
|
all lines: cytopenias, plt (storage pool dz) and NK cell dysfunction
|
|
|
other non-heme related sx associated with chediak higashi syndrom
|
- other granule-containing cells including:
- melanosomes (partial occulocutaneous albinism) - neurones- neurologic abnormalities |
|
|
how are PMNs affected in Chediak Higashi syndrome
|
reduced killing and chemotaxis - severe recurrent infxns
|
|
|
what is in the chediak higashi granules
|
MPO, SPE, AP
|
|
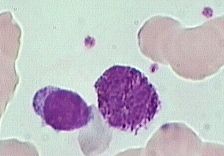
what is it
|
alder riley anomaly - looks like toxic granulation (via infection)
increased density of azurophilic granules, red lilac color; <1 um, 100 per cell |
|
|
what is the underlying defect in Alder Riley anomaly
|
mucopolysaccharidoses (Hurler, Hunter, San Fillipo, maroteauxx-Lamy, not moriquo
|
|
|
what is the inheritance pattern for alder riley anomaly
|
autosomal recessive
|
|
|
what other heme cell lines are affected in alder riley anomaly
|
lymphocytes can also contain granules - gasser cell
|
|
|
what organ systems may be affected by mucopolysaccaharidoses
|
skeletal, neuro, CT, cardiac
|
|
|
what is the alder riley anomaly composed of and what stain can you use to identify them
|
mucopolysaccharide (PAS+)
|
|
|
in what setting do you see the Pelger Huet anomaly in most PMNS
|
autosomal dominant inheritance due to a mutated lamin B receptor
|
|
|
when can you get PMN hypersegmentation (2 examples)
|
hereditary hypersegmentation, megaloblastic anemia
|
|
|
name three places where you can see lymphocyte vacuoles
|
- spielmeyer Vogt syndrome (amaurotic idiocy)
- Mannosidosis - Jordan's anomaly (also neutrophils) |
|
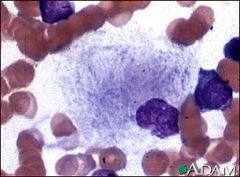
what is it
|
gaucher cell
wrinkled, striated tissue paper cytoplasm |
|
|
what is accumulating in Gaucher's disease
|
glucocerebroside
|
|
|
how is gaucher disease transmitted
|
AR
|
|
|
underlying pathology of gaucher disease
|
glucocerebrosidase deficiency (which degrades glycolipids)
|
|
|
what ethnic group is most associated with Gaucher disease
|
Ashkenazi jews
|
|
|
in what heme disease can you see an increase in pseudoGaucher cells
|
myeloproliferative syndromes (CML esp)
|
|
|
what is useful for the diagnosis of Gaucher dz
|
decreased wbc beta-glucosidase
|
|
|
what two lab findings can you see in Gaucher dz
|
- increased serum acid phosphatase
- increased serum ACE |
|
|
what heme malignancies are patients with Gaucher at increased risk for
|
MM, CLL
|
|
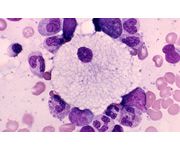
what is it
|
niemann pick dz -
foamy vacuolated cytoplasm (cf gaucher cell) |
|
|
what accumulates in niemann pick disease
|
sphingomyelin, weakly PAS+
|
|
|
in Gaucher dz, what does the gaucher cell stain for
|
acid phosphatase and PAS
|
|
|
what can you do to be sure that a niemann pick cell is that
|
-stain with PAS (wk)
- phase microscopy, pos birefringence on polarized light, yellow green on UV |
|
|
how does one diagnosis niemann pick cell
|
look for a reduced in wbc spingomyelinase
|
|
|
which has a more rapid clinical course niemann pick cell and gaucher cell
|
niemann pick dz
|
|
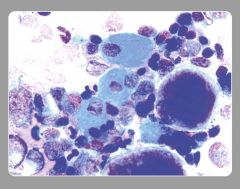
what is it
|
sea blue histiocyte
deep blue green staining granules but yellow on H&E |
|
|
what is accumulating in the sea blue histiocyte
|
ceroid
|
|
|
what is the etiology of the sea blue histiocyte
|
can be primary or secondary:
inherited (niemann-pick, wolman, fabry) secondary: MDS, CML, ITP, lymphoma, MM, parenteral nutrition |
|
|
what stains can be useful in verifying a sea blue histiocyte
|
sudan black B, Pas
also autofluoresence and characteristic EM |
|
|
what characteristic EM findings are found on Sea blue histiocytes
|
myelin bodies
|
|
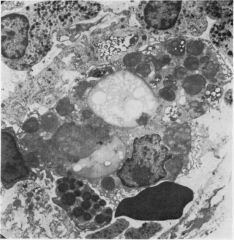
what is it
|
myelin bodies (found in sea blue histiocytes - saw in another place that can have fingerprint arrangement?)
|
|
|
what is the underlying pathology of chronic granulomatous disease
|
defect in NADPH oxidase (enzyme in respiratory burst catalyzes O2 to O2-)
|
|
|
what pathology do you find in chronic granulomatous disease
|
granulomas in multiple organs
|
|
|
what type of bacteria are those with CGD at risk for
|
catalase positive organisms (b/c catalase degrades H202 produced) and the host (who can't produce it) has no defense
|
|
|
what is the most of transmission of most cases of CGD
|
x linked but also autosomal recessive forms
|
|
|
what genes seem to be involved in causing CGD
|
phoxes (phagocyte oxidase enzymes)
|
|
|
what specific organisms are patients with CGD at risk for (5)
|
staph aureus, salmonella, serratia, aspergillus, nocardia
|
|
|
what organs are most affected in CGD
|
lung, LN and skin
|
|
|
how diagnose CGD
|
-NBT reductase negative (can't reduce NBT to insoluble purple formazan granules)
-reduced O2 consumption (after PMA stimulation) -reduced H202 and O2- production |
|
|
what is leukocyte adhesion molecule deficiency on a molecular level
|
autosomal recessive deficiency in CD11/CD18 heterodimer (they have a common B chain that is messed up)
results in leukocyte abnormalities: adhesion, chemotaxis, oxidative bursts, degranulation |
|
|
what are the clinical findings in leukocyte adhesion molecule deficiency
|
recurrent infections - Skin, ear, gums (periodontitis)
NO PUS but neutrophilia (can' t marginate!) delayed wound healing (includ umbilical cord) |
|
|
how is diagnosis of leukocyte adhesion molecule deficiency made
|
flow
functional PMN defects (reduced motility, phagocyto) |
|
|
what is the mode of transmission of myeloperoxidase deficiency
|
autosomal recessive
|
|
|
which granules of the PMN are seen on regular staining- primary or secondary
|
most that are visualized routinely are secondary neutrophilic granules (primary azurophilic granules can't be appreciated on routine staining)
|
|
|
where is MPO found in the PMN - primary or secondary granules.
|
primary (thus can't see problem on routine staining)
|
|
|
what does MPO do
|
catalyzes H2O2 to HClo- (hypochlorous acid)
|
|
|
how is MPO deficiency diagnosed
|
-automated differential (not all instruments but some)
- cytochemistry - Flow (immunophenotyping) |
|
|
are there significant infection problems in MPO deficiency
|
not so much, just in diabetics may see increased candidal infections
|
|
|
what are toxic granulations
|
look like Alder Riley anomaly (increase in density of azurophilic granules)
these are due to rapid cell division (can't dilute out granules) can be associated with toxic vacuolizaiton |
|
|
in what disease state can you see an increase in PMN vacuoles
|
MDS: 17p- syndrome
|
|
|
3 relatively specific causes of toxic granulation
|
- bacterial infection
- marrow recovery (from chemo) - hematopoietic growth factors (G-CSF, GM-CSF) |
|
|
what causes hypogranularity of PMNs
|
washed out pmns
MDS, AML |
|
|
what are Dohle bodies
|
irregular blue shaped area in cytoplasm (ribosomes, RER)
cf to May-Hegglin anomaly (Dohle bodies are smaller, less well defined and more irregular) |
|
|
causes of Dohle bodies
|
infection/BM stimulation (G-CSF)
pregnancy |
|
|
causes of PMN with hypersegmentation
|
megaloblastic anemia
MPD MDS iron deficiency |
|
|
what is the definition of PMN hypersegmentation
|
1 PMN with 6 lobes or 5% with 5 lobes (or 3 with 5 lobes)
|
|
|
causes of pseudopelgerization
|
MDS
other heme malign myxedema infections (malaria, TB) |
|
|
what are downey cells
|
activated (atypical) lymphocytes
|
|
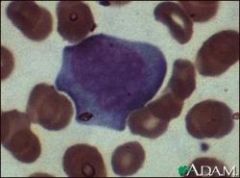
what is it
|
downey cell; low N: ratio, sometimes vacuolated cytoplasm, blast-like open chromatin
scalloped/indented edges with adjacent rbcs increased basophilia at edges (dutch skirt) |
|
|
what is the cell type that transforms into a downey cell
|
CD8 cytotoxic cell
|
|
|
causes of Downey cells (activated atypical lymphocytes)
|
- mono, EBV, CMV, HIV, toxo
|
|
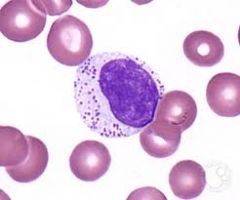
what is it
|
LGL - not pathologic per se - can have persistent elevations of theses: represent true N cells or cytotoxic T cells (need flow to distinguish)
large discrete azurophilic granules composed of perforin (punch holes in cells) and granzymes (induces apoptosis) |
|
|
what is erlichiosis
|
two tick borne diseases affecting monocytes (human monocytic erlichiosis) and granulocytes (human granulocytic erlichiosis)
|
|
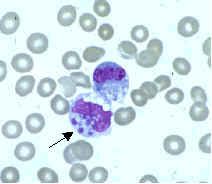
what is it
|
erlichiosis inclusion: morulae, mulberry-like structures, represents a microcolony of bacteria
|
|
|
name five general causes of hemophagocytosis
|
- viral infections (esp EBV, CMV)
- generalized immune activation - autoimmune disorders - maybe neoplastic (ex. T cell lymphomas) - maybe hereditary (ex perforin mutation) |
|
|
name two settings in which you could see emperipolesis
|
histiocytes - Rosai-dorfman
megakaryocytes - normal (possible increase in MPDs, like CIMF) |
|
|
what are hematogones and what are they often confused with
|
normally occurring precursor B cells
confused often with precursor B cell ALL |
|
|
what is considered an elevated wbc count
|
>12 x 10^9/L
|
|
|
what are the three mechanisms for neutrophilia
|
>7.5
increased mobiliziation increased survival (reduced egress into tissues) increased production |
|
|
in what inherited disease can you see neutrophilia
|
leucam (CD11/18) deficiency
|
|
|
what three infections do you see a paradoxical decrease in PMNs
|
1. typhoid
2. paratyphoid 3. brucellosis |
|
|
what is brucellosis
|
caused by brucella bacteria; from unpasteurized milk or exposure to sick animals, aka undulant fever due to afternoon fever spikes
|
|
|
what is considered lymphocytosis
|
>4 x 10^9/L
|
|
|
what is an acute bacterial infection that can lead to lymphocytosis
|
pertussis (whooping cough)
|
|
|
what is a chronic infectious cause of lymphocytosis
|
TB, syphilis, brucellosis
|
|
|
what is a parasitic cause of lymphocytosis
|
toxoplasmosis
|
|
|
name five bacterial causes of monocytosis
|
>1 x 10^9/L
TB, brucellosis, endocarditis, syphilis rickettsial (RMSF, typhus) |
|
|
name three parasitic causes of monocytosis
|
malaria, trypanosomiasis, kala-azar
|
|
|
inherited causes of eosinophilia (>.4 x 10^9/L) (just read)
|
Wiskott-Aldrich, familial reticuloendotheliosis, ectodermal dysplasia, thrombocytopenia-absent radius syndrome, congenital cardiac abnormalities
|
|
|
what is Wiskott Aldrich syndrome
|
rare X-linked recessive disease characterized by eczema, thrombocytopenia (low platelet count), immune deficiency, and bloody diarrhea (secondary to the thrombocytopenia)
|
|
|
inflammatory causes of eosinophilia (just read)
|
EM, PAN, sarcoid, Dressler's syndrome, pancreatitis, chronic active hepatitis, IBD, Churg strauss syndrome, eosinophilia myalgia syndrome
|
|
|
what is Churg Strauss
|
used to be called variant of PAN;
medium and small vessel autoimmune vasculitis, leading to necrosis. lungs (it begins as a severe type of asthma), gastrointestinal system, and peripheral nerves, but also affects the heart, skin, and kidneys. |
|
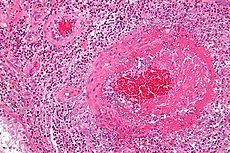
what is it
|
churg strauss: vasculitis with eosinophils, necrosis
|
|
|
what is Dressler's syndrome
|
pericarditis post MI (with pain, fever, effusion)
|
|
|
name 6 dermatopathic diseases associated with eosinophilia
|
-eczema
-psoriasis - dermatitis herpetiformis -pemphigus - pemphigoid - TEN |
|
|
what is PIE syndrome
|
pulmonary infiltrates with eosinophilia
|
|
|
what is the bug that causes Whipple's disease
|
- tropheryma whippeli
|
|
|
what inflammatory disease can have associated lymphopenia
|
sarcoid
|
|
|
what specific neoplastic condition can be associated with monocytopenia
|
Hairy cell leukemia
|
|
|
what is the way that deletions or hypermethylation can be associated with leukemia pathogenesis
|
reduce amount of a tumor suppressor gene
|
|
|
what fx, if present, is really helpful in distinguishing between lymphoblasts and myeloblasts
|
Auer rods: means myeloblast
|
|
|
in comparing lymphoblasts and myeloblasts, which are GENERALLY: larger
|
myeloblasts
(lymphoblasts are small to medium, myeloblasts would be medium to large) |
|
|
in comparing lymphoblasts and myeloblasts, which are GENERALLY: which has granules
|
myeloblasts
Lymphoblasts can rarely have granules in some cases of granular ALL, can see coarser granules in blastic NK leukemias) |
|
|
in comparing lymphoblasts and myeloblasts, which are GENERALLY: have a higher N:C ratio
|
lymphoblasts
(usually high N: C ratio); myeloblasts usually have a relatively low N:C ratio |
|
|
in comparing lymphoblasts and myeloblasts, which are GENERALLY: more clumped chromatin
|
lymphoblasts generally have more clumped chromatin, myeloblast chromatin tends to be more open
|
|
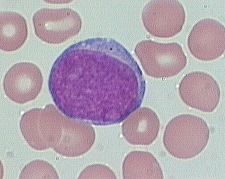
what is it
|
myeloblast (10-20m dia)
shouldn't be in peripheral blood large round to oval nucleus, fine diffuse immature chromatin (without clumping), prominant nucleolus. cytoplasm is basophilic without granules |
|
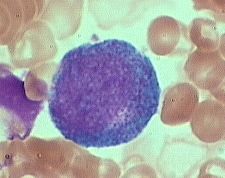
what is it
|
promyelocyte (10-20m) is slightly larger than a blast.
shouldn't be in peripheral blood slight chromatin condensation, less prominent nucleoli. cytoplasm contains striking azurophilic granules or primary granules. |
|
|
what are seen in the granules of a promyelocyte
|
These (primary) granules contain myeloperoxidase, acid phosphatase, and esterase enzymes.
|
|
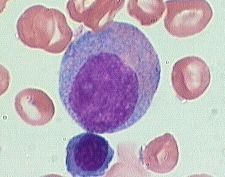
what is it
|
Myelocytes (10-18m) are slightly smaller than promyelocytes
Myelocytes are not normally found in the peripheral blood. have eccentric round-oval nuclei, often flattened along one side. The chromatin is fine, but shows evidence of condensation. Nucleoli may be seen in early stages . Primary azurophilic granules are still present, but secondary granules predominate. Secondary granules (neut, eos, or baso) first appear adjacent to the nucleus. |
|
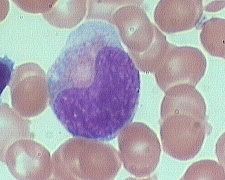
what is it
|
Metamyelocytes (10-18m) are slightly smaller than myelocytes.
really rarely in blood (0-1%) - kidney shaped indented nuclei and relatively dense chromatin, especially along the nuclear membrane. -Cytoplasm is faintly pink with almost no blue background. Numerous secondary granules (neutro, eos, or baso) clearly outnumber primary granules. |
|
|
when you see secondary granules for first time in myeloid development, what is the cell, by definition
|
myelocyte
|
|
|
in myeloid development, which is the first cell type that is "allowed" out into peripheral blood
|
metamyelocyte
|
|
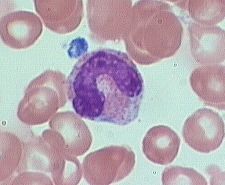
what is it
|
Bands, slightly smaller than juveniles
0-6% of peripheral blood are marked by a U-shaped or deeply indented nucleus. Opposite sides or lobes are of roughly equal size or diameter. There is no nuclear constriction > than 1/2 the lobe diameter. The chromatin is heavily clumped - secondary or specific granules in either neutrophilic or basophilic predominate |
|
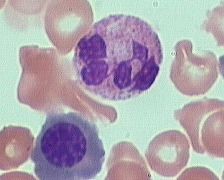
what is it
|
Segmented (segs) or polymorphonuclear (PMN) leukocytes (average 14 m dia)
45-75% of the peripheral blood white cells are segmented neutrophils. distinguished by definite lobation with thin thread-like filaments of chromatin joining the 2-5 lobes. The chromatin of the segmented neutrophil is coarsely clumped and the cytoplasm is pink due to large numbers of secondary granules. |
|
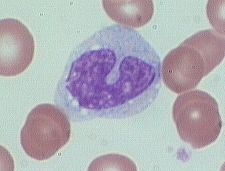
what is it
|
monocyte
monocyte is a large cell (2-3 RBC diameters). There is abundant cytoplasm with scattered fine granules against a blue-gray background. The chromatin is reticular, that is, with fine, almost linear clumping and no smudging as is seen in lymphocytes. |
|
|
are pmns phagocytic
|
yes
|
|
|
in comparing lymphoblasts vs. myeloblasts which has more prominent nucleoli
|
myeloblasts: more and more prominent
|
|
|
cytochemistry for identifying myeloblasts (5)
|
MPO+, Sudan black+, chloracetate esterase, alpha-naphthylacetate esterase, diffuse PAS
|
|
|
cytochemistry for identifying lymphoblasts
|
limited; block PAS staining
can get focal alpha-naphthylacetate esterase aNSE in T-ALL and was used for that before immunophenotyping was around) |
|
|
cytochemistry for monoblasts (4)
|
MPO, sudan black, alpha-naphthylacetate esterase (aNSE) and diffuse PAS)
|
|
|
what is the only real cytochemical difference between myeloblasts and monoblasts
|
myeloblasts are SPE + (chloracetate esterase); monoblasts are negative
|
|
|
what is MO
|
minimally differentiated AML
|
|
|
does MO AML have MPO expression
|
no, must look for characteristic granules by EM or immunophenotype (CD13, CD33 or CD117/c-kit)
|
|
|
what IHC is useful for MO AML
|
HLA-DR, CD13, CD33, CD34 and CD117, (+/-CD11c)
|
|
|
what is M1 AML
|
myeloblastic, without maturation (<10%)
|
|
|
what ihc is useful for M1 AML
|
like MO; HLA-DR, CD13, CD33, CD34 and CD117, (+/-CD11c)
but gains variable expression of CD14 |
|
|
what ihc is useful for M2
|
similar to M1 HLA-DR, CD13, CD33, CD34 and CD117, (+/-CD11c, +/-CD14) but less reliable expression of HLA-DR and CD34
|
|
|
what ihc is useful for M3
|
CD33/CD13 (others are variable)
|
|
|
what ihc is useful for m4
|
like Mo (ish)
HLA-DR, CD13, CD33, CD34 and CD117, (+/-CD11c) |
|
|
what ihc is useful for M5
|
like Mo HLA-DR, CD13, CD33, CD34 and CD117, (+/-CD11c)
except lose CD34 |
|
|
what ihc is useful for M6
|
might see HLA-DR or CD11c but the only reliable marker is totally unlike all of the AmL markers: CD235a
|
|
|
what ihc is useful for M7
|
variable expression with basic aml markers: HLA-DR, CD34 and CD33 but add in CD41
|
|
|
what is M2 AML
|
myeloblastic, with maturation (>10%)
|
|
|
what cytogenetic finding is seen in M2
|
t(8;21) - associated with good prognosis
AML1-ETO |
|
|
what is M3
|
promyelocytic AML, (APL); there is a M3v which is a microgranular variant of APL
|
|
|
what is the cytogenetic abnormality of M3 Aml
|
t(15;17); RARa-PML - generally considered good prognosis
|
|
|
what clinical fx are relatively distinct for M3 form of AML
|
bleeding diathesis (DIC)
|
|
|
what morphologic fx are distinctive about M3 AML
|
bundles of Auer rods (=faggots)
cottage loaf nuclei |
|
|
what is M4 AML
|
myelomonoblastic AML
|
|
|
what are the cytogenetic findings in M4
|
11q23 MLL
|
|
|
what is M4Eo
|
myelomonoblastic with abnormal eosinophils
|
|
|
what are the cytogenetic findings in M4Eo
|
inv(16), CBFbeta-MYH11
good prognosis cytogenetic finding |
|
|
what are the three good prognosis cytogenetic findings in AML
|
-inv(16)
t(15;17) t(8;21) |
|
|
what is M5
|
monoblastic AML (without A and with maturation B)
|
|
|
what cytogenetic findings are seen in M5 aml and what is this similar to
|
11q23 MLL (like M4 but not M4Eo)
|
|
|
what clinical findings are seen in M4 and M5
|
tissue infiltration with skin, gums, CNS involvement
|
|
|
what is m6
|
erythroblastic (blasts with >20% nonerythroid cells)
>50% rbc precursors in marrow |
|
|
what is m7
|
megakaryoblastic aml
|
|
|
what cytogenetic findings are in m7
|
3q21q26
t(1;22) OTT-MAL |
|
|
what are useful immunophenotyping specifically for M7
|
CD61, CD41, CD42, vWF
|
|
|
when is immunophenotyping in Aml most useful
|
only at the diagnosis of the two "extremes" Mo and M7
|
|
|
what is the story of CD33 in the context of aml
|
Mo CD33 - early myleomono-restricted ag, expression decreases with maturation
|
|
|
what is the story of CD13 in AML
|
Mo, aminopeptidase N, coronavirus receptor
|
|
|
what is the story of CD117 in AML
|
ckit, stem cell facotr recptor, perhaps more specific than CD33/cd13 in the diagnosis of MO
|
|
|
what is the story of CD61 in AML
|
useful for M7; gpIIIa, earliest antigen expressed in megakaryoblastic development
|
|
|
what is the story of CD41 in AML
|
m7; gbIIb/IIIa - fibrinogen receptor (abnormal in Glanzmann disease)
|
|
|
what is the story of CD42 in AML
|
in M7; gbIb/IX; vwF receptor; abonomal in Bernard Soulier syndrome
|
|
|
what is hte story of CD36 in AML
|
in M7; gpIIIb/IV, thrombospondin receptor (also in M4, M5, M6)
|
|
|
generalizations with phenotyping in AML: what two antigens does M3 (APL) often lack
|
CD34 and HLADR (both blast ags)
|
|
|
what are the 6 ihc that are associated with monocytic maturation and which one is the "best"
|
CD4, CD11b, CD14, CD24, CD36, CD64 (CD64 may be the best)
|
|
|
what percent of AML can have tdt
|
15%
|
|
|
what is the usual order of ihc molecules in myelopoiesis (5)
|
CD34 -> CD117 -> CD33 -> CD13 -> CD15
|
|
|
be aware that some true myeloid (not mixed lineage) leukemias can have more classically associated lymphoid markers (read examples)
|
M2 - CD19
M3v and M4Eos are CD2 many express T cell antigens (CD7>CD2 and CD5) |
|
|
what is the current classification (WHO) for grouping AML
|
1. AML with recurrent cytogenetic abnormalities
2. AML with multilineage dysplasia 3. AML and MDS/therapy related 4. AML, NO catergorized 5. AML of ambiguous lineage |
|
|
what are the AMLs with recurrent cytogenetic changes
|
1 AML with t(8;21), AML1 (CBFa/ETO)
2 APL with t(15;17) and variants (PML/RARa) 3 AML with abnl bone marrow eos (inv(16) or t(16;16) - CBFb/MYH11 4 AML with 11q23 (MLL) |
|
|
what are the two therapy related AML/MDS types
|
alkylating agent related
epipodophyllotoxin related (have more 11q23 translocations) |
|
|
what are most ALL positive for (>95%)
|
Tdt
|
|
|
what does Tdt
|
nipples and adds "n" regions at time of antigen receptor gene rearrangement (to generate more diversity in both igs and tcrs)
|
|
|
what are pan B cell antigens - 4 relatively specific and two that are helpful but nonspecific
|
4 more specific: CD19, CD20, CD22, CD79a
2 more nonspecific: CD24, HLA-DR |
|
|
b maturation antigens in order
|
Tdt -> CD10 -> Cu -> SmIg (note: CD20 is also a maturation antigen)
|
|
|
pan t cell antigens (including some nonspecificity)
|
CD2, CD5, CD7 and later cCD3 (recall: CD5 can be found on some B cell subsets, CD7 on myeloid precursors, some AMl also CD2 and CD5)
|
|
|
t maturation antigens
|
Tdt -> CD1a and CD4/CD8 coexpression -> sCD3 and CD4 or CD8
|
|
|
where can you find CD10+ (6 examples, but several others)
|
precursor B cell ALL; germinal center cells, mature PMNs, stage II thymocytes, breast, renal)
|
|
|
what immunophenotype what might you expect on precursor B cell ALL
|
b-ag+, CD10+, SmIg-
|
|
|
pre B ALL associated with t(1; 19)
|
B-Ag+, Cmu, CD34-
|
|
|
what is t(1;19) in preB ALL
|
E2A: PBX1 fusion
|
|
|
t cell lymphoblastic lymphoma immunophenotype
|
T -Ag+, CD1a, CD4, CD8 (may also be CD10)
|
|
|
pre B ALL associated with t(9;22) in adults
|
CD13, CD33
|
|
|
what is pro bALL
|
Pro-B-ALL, one of the most immature ALL subtypes, is characterized by the expression of cytoplasmic (cy) or membrane CD22, CD19, CD24, and CD79a, and by the absence of CD10 and of cy or surface immunoglobulins
|
|
|
profile of two antigens (1 positive, 1 neg) in pro B ALL that are thought to be predictive of t(4;11)
|
CD15+, CD10-
|
|
|
what is the story of t(4;11) in pro B-ALL
|
worse prognosis, found most in the <12 months of age with ALL
|
|
|
good prognosis in ALL
|
1) t(12;21) - most common ped abnormality (25) need molecular or FISH to demo TEL/ETV6:AML1 fusion
2) hyperploidy >50 czomes |
|
|
bad prognosis in ALL
|
1. t(9;22) - alsob bcr abl+ but different bcr breakpoint, predominantly in adult ALL
2. t(1;19) - poor prognosis in pre B ALL unless with tailored therapy 3. t(4;11) - associated with neonatal acute mixed lineage leukemia 4. t(8;14) associated with L3, IGH-MYC - variants include t(2;8) - kappa; t(8;22) lambda 5. hypodiploidy |
|
|
three different clinical scenarios involving 11q23 translocations (disrupting MLL gene)
|
- acute myelomonoblastic leukemia (M4, M5)
- acute myeloblastic leukemia due to chemo (epipodophyllotoxins) - neonatal acute (mixed lin) leukemias |
|
|
most common molecular abnormality in AML
|
FLT3 mutations (internal tandem duplication or point mutation)
|
|
|
are FLT3 mutations good or bad
|
poor prognosis
|
|
|
three mutations in ALL with prognostic relevance
|
CEPBA, CKIT, NPMI
|
|
|
characteristics of myeloproliferative disorders (9 fx) try to memorize as much as possible
|
- clonal stem cell d/o
- increased # circulating matures/maturing nonlymphoid cells - committment (not absolute) to one lineage - splenomegaly - extramedullary hematopoiesis - myelofibrosis - some functional abnormalities of mature cells - variable tendency to evolve/transform to acute leukemias - increased basophils |
|
|
which myeloproliferative d/o is most likely to evolve into an acute leukemia
|
CML
|
|
|
what is the myeloproliferative d/o that involves: granulocytes
|
CML
|
|
|
what is the myeloproliferative d/o that involves: erythrocytes
|
polycythemia vera
|
|
|
what is the myeloproliferative d/o that involves: platelets
|
Essential thrombocythemia
|
|
|
compare the differential count of granulocyte precursors in leukemoid reaction vs. CML
|
leukemoid reaction: increasing amounts of blasts, pros, myelos, metas, polys
CML: increasing amounts of blasts, pros, myelso, metas and polys but "spiked" levels of myelos and polys (myelo bulge) and mature polys increased |
|
|
what are the various stages of myeloid precursors
|
blasts - pros - myelos- metas - polys
BPMMP |
|
|
what is the LAP score
|
leukocyte alkaline phosphatase - cytochemical test on peripheral smears designed to distinguish between leukemoid reaction (high) vs. CML (low). PNH is also low
|
|
|
where are toxic granulations seen: leukemoid reaction or CML
|
leukemoid reaction
|
|
|
where are basophils more likely to be seen: leukemoid reaction or CML
|
CML
|
|
|
what age targets CML
|
~50 years
|
|
|
how high is the wbc count in CML
|
usually in the >50K, often >100K
|
|
|
what happens to hemoglobin in CML
|
mildly decreased
|
|
|
which is the ph czome itself
|
shortened 22q-
|
|
|
if "forced" to do a western blot for bcr-abl gene product, what "gene product" would that be
|
p210
|
|
|
which is more critical to the diagnosis of CML - peripheral blood or bone marrow
|
peripheral blood is where the diagnosis is made
|
|
|
what does the bone marrow look like in CML
|
- HYPERcellular
- pseudoGaucher cells (from increased cell turnover) +/- fibrosis |
|
|
what time frame do most with CML evolve into acute leukemia
|
3-5 years
|
|
|
what additional cytogenetic abnormalities can be seen in blast crisis for CML
|
(Ph, i(17q) and 8)
|
|
|
if CML were to undergo a lymphoblastic crisis vs. myeloblastic crisis, which would have a better prognosis
|
30% lymphoblastic (better prognosis)
70% myeloblastic (worse prognosis) |
|
|
if there is no Ph czome, what is a CML-like disease called
|
consider CMML or atypical CML
|
|
|
what is the new name for juvenile CML
|
renamed JMML; unusual and quite aggressive pediatric disease
|
|
|
what is JMML
|
the new name of juvenile CML; aggressive pediatric dz; increased HbF; hypersensitive to GM-CSF in vitro
|
|
|
what mutations are seen somewhat in JMML
|
NF1, PTPN11 and RAS mutaitons
|
|
|
what is polycythemia vera
|
neoplastic proliferation and maturation of erythroid, megakaryocytic and granulocytic elements to produce what is referred to as panmyelosis.
|
|
|
what is the molecular mechanism underlying PV
|
mutation in Jak2 pathway resulting in altered EPO receptor and causes cells to be hypersensitive to EPO.
|
|
|
what is a major difference between those with PV and those with secondary erythrocytosis
|
In contrast to secondary polycythemias, PCV is associated with a low serum level of the hormone EPO
|
|
|
what is Monges disease
|
increase in red blood cells due to chronic hypoxia due to chronic living in high altitudes
|
|
|
what is Pickwickian syndrome
|
obesity -> sleep/obstructive apnea -> chronic hypoxia -> increased red cells
|
|
|
name three Hb in which O2 affinity is altered
|
high O2 affinity Hb (Chesapeake)
metHb COHb |
|
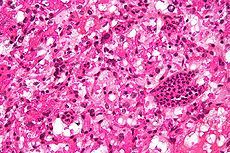
what is it and what is it's association with secondary polycythemia
|
cerebellar hemangioblastoma; VHL mutations result in failure to degrade hypoxia-inducible factor 1 that stimulates EPO and other hormones resulting in tumor growth as well as secondary polycythemia. RELATED TO VHL
|
|
|
what is Gaisbock syndrome and how does it relate to discussion of polycythemias
|
occurs in sedentary obese males with high calorie diet; results in reduced plasma volume and relative increase in red blood cells (like dehydration would do too)
|
|
|
demographics and sx of polycythemia vera
|
older age group; >60 years, sx of increased Hct (>55)
|
|
|
what two criteria are essential, but not fully sufficient, for the diagnosis of polycythemia vera
|
1. elevated rbc mass >25% above mean or Hb>18.5 in men, 16.5 in women
2. exclude secondary causes of polycythemia and no familial erythrocytosis (no increased EPO due to hypoxia, high oxygen affinity Hb, truncated EPO receptor, inappropriate EPO production by tumor |
|
|
"softer" diagnostic criteria for evaluation of polycythemia vera (just read through)
|
in addition to elevated rbc mass/hb and exclusion of other causes:
- splenomegaly - clonal genetic abnormality that is not 9;22 - endogenous erythroid colony formation in vitro - thromboycytosis >400, wbc>12, - BM bx with panyelosis and prominent erythroid/megakaryocytic proliferation - low serum EPO levels |
|
|
what is endogenous erythroid colony formation in vitro
|
?? using bone marrow cells in culture, cells from patients with PV should be able to produce erythroid colonies in the absence of exogenous EPO
|
|
|
some additional clinical fx of PV
|
- often iron deficient
- live for 10-15 years - morbidity/mortality from hemorrhage/thrombosis - acute transformation is unusual |
|
|
two mutations that might be found in PV
|
PRV1 and Jak2
|
|
|
what are important fx of myelofibrosis
|
- massive splenomegaly (extramedullary hematopoiesis, also in liver, LN)
- leukoerythroblastic picture in peripheral blood - marked fibrosis on BM bx (dry tap) - no t(9;22), no bcr-abl |
|
|
what age does myelofibrosis target
|
older ~65 years
|
|
|
how are different cell lines affected in blood
|
- wbc - may nl, increase, decrease
- plt - bizarre - may have pancytopenia |
|
|
what is a relatively specific sign for myelofibrosis compared to other MPD
|
elevated levels of circulating CD34 cells
|
|
|
what mutation might (~50%) be found in myelofibrosis
|
Jak2
|
|
|
what is the cause of fibroblasts in marrow?
|
not neoplastic, secondary due to elaboration of growth factors (PDGF, TGFb, bFGF)
|
|
|
what is the morbidity/mortality in myelofibrosis related to (3)
|
hemorrhage, thrombosis, hypersplenism
|
|
|
how common is acute transformation in myelofibrosis
|
unusual ~15%
|
|
|
what age group is essential thrombocythemia occur in
|
older ~60 years
|
|
|
what is the least common myeloproliferative d/o
|
essential thrombocythemia
|
|
|
what are the criteria for diagnosis of ET
|
not all monoclonal, even allow bcr-abl in some
platelet count of >600K exclude secondary causes of thrombocytosis |
|
|
name three causes of secondary thrombocytosis
|
chronic inflammation
asplenia iron deficiency |
|
|
what is HUMARA-XCIP assay
|
it is a way of assessing clonality based of x czome inactivation evaluation
|
|
|
if a case of ET is not monoclonal, how is the clinical course different
|
less thrombosis
|
|
|
what other cell line changes are seen in ET
|
abnormal megakaryocytes with clustering, in marrow (something circulate)
|
|
|
acute transformation rates in ET
|
<<5%
|
|
|
can jak2 mutations occur in ET
|
~50% (ET and myelofibrosis are the same in this way)
|
|
|
list of "other" myeloproliferative d/o that are rare (5 listed; three are hybrid mds/mpd)
|
- chronic neutrophilic leukemia - may have t(9;22) but involves 3'mu-bcr(e19) with p230
- chronic eosinophilic leukemia/hypereosinophilic syndrome (FIP1L1-PDGRFRA) - **atypical chronic myeloid leukemia **juvenile myelomonocytic leukemia **chronic myelomonocytic leukemia **hybrid MDS/MPD |