Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
55 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Rate-limiting step of de novo purine synthesis:
|
Glutamine-PRPP amidotransferase
|
|
|
|
Rate-limiting step of Urea cycle
|
Carbamoyl phosphate synthetase I
|
|
|
|
Rate-limiting step of Fatty acid synthesis
|
Acetyl-CoA carboxylase
|
|
|
|
Rate-limiting step of Fatty acid oxidation
|
Carnitine acyltransferase I
|
|
|
|
What's the difference between the actions of biotin and folic acid?
|
Biotin is a cofactor in carboxylation reactions -- it adds CO2
THF is methyl group donor -- it adds CH3 |
|
|
|
What is the difference between the activity of NADH and NADPH?
|
NADH is usually involved in catabolic processes, i.e. Glycolysis and the TCA
NADPH is usually involved in anabolic processes, like steroid and fatty acid synthesis. |
|
|
|
HMP Shunt:
(1) What does it do? (2) Describe the most prominent associated pathology. |
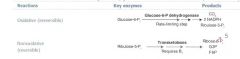
NADPH is made by the HMP shunt, AKA the pentose-5-phosphate pathway.
A small fraction of the Glucose-6P that's made in the first step of glycolysis gets shunted off to this pathway to restore NADP+ to NADPH. |
G6PD is the rate-limiting step of the shunt. When it is defective, NADPH deficiency occurs which is minor in most tissues, but not in the erythrocytes, where the shunt is the only source of NADPH.
Result: oxidative/radical damage to RBCs and hemolytic anemia. |
|
|
How do hexokinase and glucokinase differ?
(1) General description (2) Kinetics |
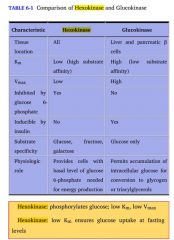
Both of these enzymes phosphorylate glucose as part of the first step of glycolysis.
Hexokinase is found in all cells and specializes in operating in a low glucose environment to provide a small amount of glucose for each cell's needs. Glucokinase is found in the liver, and specializes in working in a hi glucose environment to rapidly phosphorylate lots of glucose, providing glycogen storage for the whole body. and is inhibited by the presence of glucose 6-P. This allows individual cells to burn only as much glucose as they need. Glucokinase is found in the liver, and is not; this allows the organ to function as a glycolytic machine for the rest of the body. |
Hexokinase has a hi affinity for glucose and a LOW K_m, so it can operate in low glucose environments.
It also has a low V_m, so that regardless of the environment, it doesn't make a lot Glucokinase has a LOW substrate affinity and a HI K_m, but a very hi V_max, so it works best where there's lots of glucose, and makes stuff for the whole body. |
|
|
(1) What is the rate-limiting step of glycolysis, and (2) how is it regulated?
|

Fructose-6P + ATP --PFK-1--> Fructose-1,6-bisphosphate
|
As the rate-limiting enzyme, PFK-1 is:
Downregulated by ATP, Citrate Upregulated by AMP, fructose-2,6-BP |
|
|
Which two steps of glycolysis consume ATP?
|

Glucose --Gluco/Hexokinase--> G-6P
Fructose-6P --PFK-1--> Fructose-1,6-bisphosphate |
|
|
|
In which two steps does glycolysis produce ATP?
|
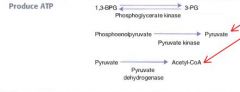
#1: yields 2 ATP
1,3-bisphosphoglycerate --phosphoglycerate kinase--> 3-phosphoglycerate #2: also yields 2 ATP Phosphoenolpyruvate--Pyruvate kinase--> Pyruvate |
|
|
|
How do blood sugar levels interact with the rate-limiting step of glycolysis?
(main idea) |
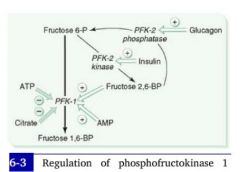
Background:
2 enzymes act on the substrate of hte rate-limiting reaction: PFK-1 and PFK-2 PFK-1 is responsible for the main substrate. **In the presence of insulin, PFK-2 is activated. **PFK2's activity effectively induces PFK-1 (and therefore glycolysis). |
|
|
|
(1) What is the mechanism by which insulin interacts with the rate of glycolysis?
(2) How about glucagon? |
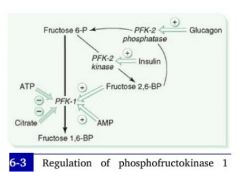
Hi blood sugar --> hi insulin --> decreased cAMP --> dephosphorylation of PFK-2 gives it a kinase activity --> increased production of Fructose-2,6-bisphospate --> increased PFK activity --> increased glycolysis.
|
Low blood sugar --> hi glucagon --> hi cAMP in cells --> PFK-2 is phosphorylated and given a phosphatase activity --> PFK-2 reconverts Fructose-2,6-bisphosphate to fructose-6P --> Fructose-2,6-Bisphosphate levels decrease and PFK-1 is downregulated --> Glycolysis stops.
|
|
|
What cofactors are required for pyruvate processing enzymes to function?
|

1. Pyrophosphate (Thamine/TTP)
2. FAD (B2, riboflavin) 3. NAD (B3, niacin) 4. CoA (B5, pantothenate) 5. Lipoic Acid |
We treat the enzyme complex responsible for pyruvate processing as though it were a single enzyme, pyruvate dehydrogenase, but obviously it's 3 enzymes. Don't worry about them.
|
|
|
Pyruvate Dehydrogenase Deficiency
Etiology & Presentation |

Causes:
1. Genetic deficiency 2. Cofactor deficiency (i.e. B1 def in alcoholism) |
Presentation:
Neurological defects |
|
|
So we've made pyruvate from glucose. What are the 4 things we can do with it?
|

Can be made into:
1. A.Coa for the TCA 2. Lactic acid fermentation 3. Oxaloacetate for either Gluconeogenesis or TCA 4. We can package waste NH3 from the periphery into it, turning it into alanine. [This is then shipped back to the liver for entry into the Urea Cycle.] |
|
|
|
What are the products of one turn of the TCA Cycle?
|
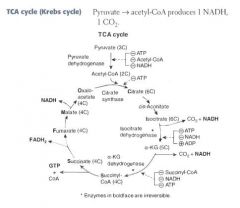
For each turn of the cycle, we burn 1 A.CoA. The net output is:
3 NADH 1 FADH2 1 GTP 2 CO2 = 12 ATP |
|
|
|
What are the enzymes of the TCA cycle?
|
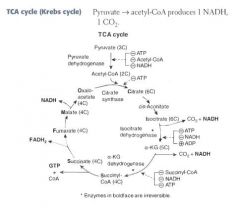
Citrate Is Krebs Starting Substrate For Making Oxaloacetate!!
Citrate Isocitrate α-ketoglutarate Succinyl CoA Succinate Fumarate Malate Oxaloacetate |
|
|
|
Where and how is α-ketoglutarate metabolized?
|
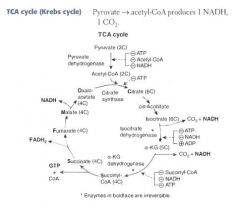
α-ketoglutarate is a metabolite of the TCA cycle:
α-KG --α-KG dehydrogenase--> Succinyl CoA |
α-ketoglutarate dehydrogenase is interesting because it is just like pyruvate dehydrogenase!
It's a complex of 3 enzymes with the same 5 cofactors: B1, B2, B3, B5, lipoic acid. |
|
|
How does arsenic affect cellular metabolism?
|

Arsenic inhibits lipoic acid from acting as a cofactor in pyruvate dehydrogenase.
The result is neurological problems, lactic acidosis |
|
|
|
Arsenic poisoning
Presentation: |
This type of toxicity presents with vomitting, rice-water stools, and garlic breath.
|
|
|
|
How does NADH made in the cytoplasm during glycolysis get restored to NAD+, so glycolysis can continue?
|
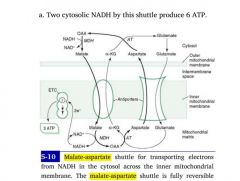
NAD+ and NADH CANNOT cross the inner mitochondrial membrane!
Instead, oxaloacetate is converted to a non-TCA substrate -- aspartate -- which crosses through a channel. |
1. Oxaloacetate --> Aspartate
2. Aspartate crosses opposite glutamine 3. Aspartate is reconverted to oxaloacetate in the cytoplasm 4. OAA accepts H:- from NADH, becoming malate 5. Malate crosses back into matrix to be reused by TCA |
|
|
List the basic amino acids.
|
Lysine
Arginine Histidine (all positively charged in serum) |
|
|
|
List the acidic amino acids.
|
Aspartate
Glutamate (both negatively charged in serum) |
|
|
|
Histone Structure
|
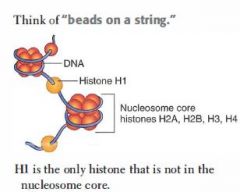
Tetramers of H2A, H2B, H3, and H4 proteins form nucleosome core.
DNA wraps twice around each core. H1 proteins are not in the nucleosome core, but rather tie the nucleosome beads together. |
|
|
|
What are the two functions of methylation DNA methylation?
|

(1) Template strand cytosine and adenine residues are methylated, indicating to DNA polymerase which strand is the template for repairs.
(2) Hypermethylation: CpG islands of DNA tend to become hypermethylated, inactivating transcription. -- CpG islands are associated with the promoters for housekeeping genes that we want to express at low basal rates. |
|
|
|
Role of acetylation in gene expression.
|
This modification, performed by histone acetyltransferases (HATs), loosens DNA from heterochromatin to euchromatin, increasing transcription.
|
|
|
|
Distinct characteristics of the 5 nucleotides:
|
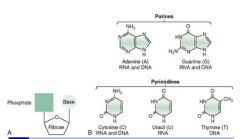
Cytosine: ammonia group
Uracil: ketone Thymine: methylated Adenine: ammonia group Guanine: ketone + ammonia group |
|
|
|
What simple pyrimidine modification is frequently responsible for mutations, and how is it repaired?
|
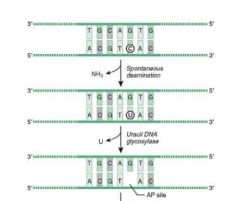
Cytosine deaminates to uracil spontaneously and at a constant rate.
-- C ==> U, resulting in an AT mismatch -- Repaired by BER |
|
|
|
How do healthy cells combat skin cancers?
|
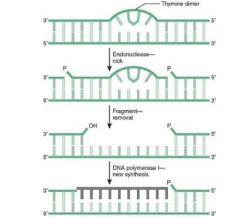
This mutation is combated with Nucleotide Excision Repair.
1. In NER, excision endonuclease detects the dimer b/c the shape is distorted, and nicks the damaged strand on both sides of the lesion, perhaps several bases down. 2. Exonucleases remove the fragment 3. DNA Polymerase I replaces the missing fragment, and DNA Ligase ligates it into the strand. |
Thymidine dimer formation is the most common cause of skin cancers, i.e.:
-- Melanoma -- Basal cell carcinoma -- Squamous cell carcinoma |
|
|
What simple purine modification is responsible for mutations, and how does it happen?
|
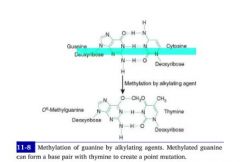
Methylation of Guanine by alkylating agents, i.e. cytoxan, results in it's mismatch with thymine.
-- G-->C -- met-G-->T |
|
|
|
State Mechanism of de novo purine synthesis. Note requirements for production of both bases.
|

1. Ribose 5-phosphate --PRPP Synthetase--> PRPP
2. PRPP + NH3 from glutamine ---> 5-Phosphoribosylamine -->-->--> Inosine monophosphate (IMP) 3a. IMP + aspartate & GTP ---> AMP 3b. IMP + NAD & ATP --> GMP |
**Guanosine requires ATP and NAD+ for its generation.
**Adenosine requires GTP for its generation. |
|
|
What is the committed step of de novo purine synthesis?
|
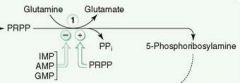
PRPP + NH3 from glutamate ---> 5-ribosylamine + PPi
|
|
|
|
What is the rate-limiting step in de novo purine synthesis?
|
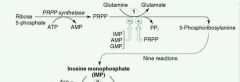
Ribose-5-phosphate ---PRPP Synthetase--> PRPP
|
|
|
|
What is the major difference between purine and pyrimidine synthesis?
|
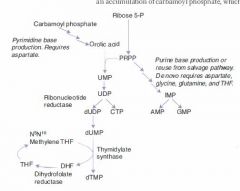
In purine synthesis we start with a ribose backbone and build a base to it.
-- *Inosine* In pyrimidine synthesis we make a temporary base and add the backbone to it, then modify the base. -- **Orotic Acid** |
|
|
|
What is the major difference between purine and pyrimidine synthesis?
|
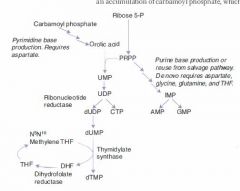
In purine synthesis we start with a ribose backbone and build a base to it.
-- *Inosine* In pyrimidine synthesis we make a temporary base and add the backbone to it, then modify the base. -- **Orotic Acid** |
|
|
|
Hydroxyurea
MoA & Indications |
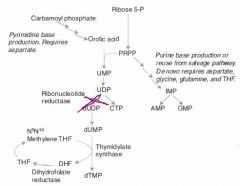
Inhibits thymine production by inhibiting ribonucleotide reductase
|
Indications:
Chemotherapeutic agent used to keep white count low in AML. |
|
|
Hydroxyurea
MoA & Indications |
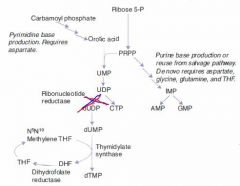
Inhibits thymine production by inhibiting ribonucleotide reductase
|
Indications:
Chemotherapeutic agent used to keep white count low in AML. |
|
|
6-mercaptopurine
MoA and Ind |
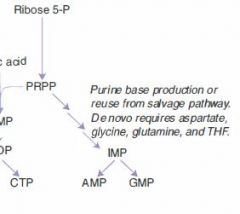
A false purine analogue that both:
(1) inhibits production of real purines via feedback inhibition (2) gets incorporated into DNA, terminating DNA synthesis. |
Maintenance therapy for children with ALL.
Often given as azothioprine, a precursor. |
|
|
6-mercaptopurine
MoA and Ind |
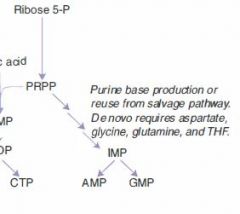
A false purine analogue that both:
(1) inhibits production of real purines via feedback inhibition (2) gets incorporated into DNA, terminating DNA synthesis. |
Maintenance therapy for children with ALL.
Often given as azothioprine, a precursor. |
|
|
5-fluorouracil
MoA & Ind |
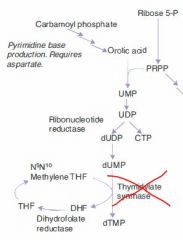
Tumor cells convert it to a false fdUMP, which irreversibly inhibits thymidylate synthase.
This blocks thymidine synthesis, resulting in a thymidine-less death. |
|
|
|
Mechanism of Pyrimidine Synthesis:
|
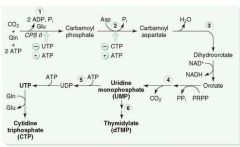
1a. As with purine synthesis, we make PRPP from Ribose-5-phosphate.
1b. CO2 + Gln + ATP ---CPT II---> Carbamoyl Phosphate 2. Carbamoyl Phosphate -->-->--> Orotic Acid 3. Orotic Acid + PRPP ---> UMP 4a. UMP ---> dUMP (or fdUMP in chemo) 4b. UMP ---ribonucleotide reductase---> CTP 5a. dUMP ---Thymidylate synthase + met-THF---> dTMP |
|
|
|
Causes of orotic aciduria.
|
(1) Inability to convert orotic acid to UMP caused by autosomal recessive deficiency in either:
-- orotic acid phosphoribosyltransferase -- orotidine 5'-phosphate decarboxylase |
|
|
|
Presentation of orotic aciduria
|
Symptoms seen in infancy:
1. Increased orotic acid in urine 2. Megaloblastic anemia 3. Failure to thrive |
|
|
|
Clinical findings in orotic aciduria:
|
Symptoms seen in infancy:
1. Increased orotic acid in urine 2. Megaloblastic anemia 3. Failure to thrive |
|
|
|
What is the committed step of pyrimidine synthesis?
|
Gln + CO2 + 2ATP ---CPT II---> Carbamoyl phosphate
The production of carbamoyl phosphate is the committed and rate-limiting step of pyrimidine synthesis. |
|
|
|
2 Purine Breakdown Pathways:
|

(1)
1. AMP ---AMP deaminase---> IMP 2. IMP ---> Inosine 3. Inosine ---> Hypoxanthine 4. Hypoxanthine ---Xanthine oxidase---> Xanthine 5. Xanthine ---Xanthine oxidase---> Uric Acid (2) 1. AMP ---> Adenosine 2. Adenosine ---Adenosine Deaminase---> Inosine 3, 4, 5: same as above |
|
|
|
SCID
Path & Presentation: |
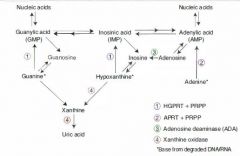
Adenosine deaminase (ADA) deficiency results in:
1) Buildup of ATP b/c we can't break it down. ATP buildup downregulates ribonucleotide reductase, resulting in decreased DNA synthesis. 2) inability to break down adenosine into inosine. Buildup of adenosine is toxic to B and T lymphocytes. |
Presentation: Bubble boy
|
|
|
Lesch-Nyhan syndrome
Path & Presentation: |
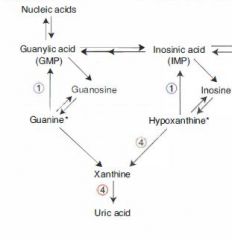
Path:
Caused by X-linked Recessive deficiency of HGPRT, which converts both hypoxanthine and guanine to IMP. Results: (1) Hypoxanthine buildup ---> excess uric acid production (2) Guanine buildup ---> excess uric acid production. |
Presentation:
MR, aggression, self-mutilation, choreoathetosis, hyperuricemia and gout. |
|
|
DNA Polymerase I
|
Prokaryotic only. Degrades RNA primer and fills gaps.
|
|
|
|
DNA Polymerase III
|
*Prokaryotes only*. Elongates leading strand by adding deoxynucleotides until it reaches primer of preceding fragment, then falls off.
|
|
|
|
What pathology results from loss of nonhomologous end joining?
|
Ataxia telangiectasia: disease consisting of cerebellar ataxia, dilation of blood vessels in skin and eyes, and immunodeficiency of T and B cell origin.
|
|
|
|
Which carbon on an incoming nucleotide bears the triphosphate?
|
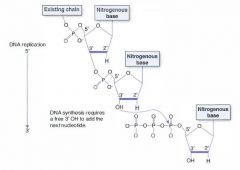
The 5' carbon bears the triphosphate.
The 3' carbon has the hydroxyl group on which the next nucleotide is attached. |
|
|
|
Stop and Start Codons:
|
Start: AUG (codes for methionine)
End: UGA U Go Away UAA U Are Away UAG U Are Gone |
|
|
|
What is the role of promoters and enhancers?
|
Promoters are sequences of DNA upstream from that are bound by RNA Polymerase and other TF's.
Enhancers are DNA sequences that bind TFs to enhance transcription. *They may be upstream, downstream, or IN an INTRON of the gene they enhance!* |
|