Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
88 Cards in this Set
- Front
- Back
|
How are most inborn errors of metabolism inherited?
|
- Autosomal recessive OR
- X-linked recessive |
|
|
What kind of defect usually causes an inborn error of metabolism? What does this lead to?
|
Single gene defect causing significant block in a metabolic pathway, causing:
- Cellular intoxication - Energy deprivation - Mixture of the two |
|
|
What can cause neurodevelopmental disability or death in a patient with an inborn error of metabolism?
|
- Early acute illness
- Repeated episodes |
|
|
What would make you suspicious that a newborn has an inborn error of metabolism?
|
– Rapid deterioration in an otherwise well infant
– Recurrent emesis or feeding difficulty – Alterations in respirations • Tachypnea, apnea • Intractable hiccups – Changing mental status/lethargy/seizures – Abnormal urine/body smell |
|
|
What would make you suspicious that infant (months to years of life) has an inborn error of metabolism?
|
– Failure to thrive
– Developmental delay – Regression in milestones – Dietary aversion: proteins, carbs |
|
|
What is the differential diagnosis for an otherwise healthy newborn who is rapidly deteriorating, may have recurrent emesis or feeding difficulty, alterations in respirations, change in mental status/lethargy/seizures, and/or abnormal urine/body smell?
|
** Sepsis
- Congenital heart disease - Pyloric stenosis - Adrenal insufficiency (eg, 21-hydroxylase deficiency) - Inborn error of metabolism |
|
|
What historical facts do you need to ask about in a newborn you think may have an inborn error of metabolism?
|
- Catabolic state induction (recent infection? fasting? dehydration?)
- Change or addition to diet (protein, carbs, fats) - Ask about consanguinity - Family history of SIDS? |
|
|
If an infant smells musty or mousy, what diagnosis do you think of?
|
Musty or Mousy
|
|
|
If an infant smells boiled cabbage, what diagnosis do you think of?
|
Tyrosinemia or hypermethioninemia
|
|
|
If an infant smells of maple syrup, what diagnosis do you think of?
|
Maple syrup urine disease
|
|
|
If an infant smells like sweaty feet, what diagnosis do you think of?
|
Isovaleric acidemia or glutaric acidemia type II
|
|
|
If an infant smells like cat urine, what diagnosis do you think of?
|
Multiple carboxylase deficiencies (Biotin deficiency)
|
|
|
What labs do you get in evaluating a sick newborn you think might have an inborn error of metabolism?
|
In general:
• Complete blood count and differential • Blood, Urine, and CSF culture (hold tubes for future) • Plasmaelectrolytes • Blood glucose • Blood gas • Call for Newborn State Screen results Specific to inborn error of metabolism: • Free flowing plasma ammonia • Lactate and pyruvate • Urineketones • Urinereducing substances • Plasma amino acids • Urine organic acids |
|
|
Who should you consult with when evaluating a sick newborn you think might have an inborn error of metabolism?
|
Genetics / Metabolic consultation
|
|
|
What are the three most important lab values to look at to see if a newborn error of metabolism is more likely?
|
- Acid base balance (metabolic acidosis)
- Ammonia level (hyperammonemia) - Glucose (hypoglycemia) |
|
|
When do you calculate an anionic gap? How?
|
If patient has metabolic acidosis
Na+ - (Cl- + HCO3-) Normal = 8-16 mEq/L Anion gap >16 mEq/L |
|
|
What are the characteristics of a metabolic acidosis?
|
- pH <7.35 (excess H+)
- HCO3- deficit - Compensatory hyperventilation (↓ PaCO2) (Need to calculate anion gap) |
|
|
What are the causes of metabolic acidosis with a normal anion gap? Treatment?
|
Chloride increased = HCO3- wasting
- GI (diarrhea) or renal tubular acidosis - Responds to giving NaHCO3 |
|
|
What are the causes of metabolic acidosis with an ↑ anion gap?
|
- Lactic acidosis
- Ketoacidosis - Organic acidopathy |
|
|
What is the definition of hypoglycemia?
|
Blood glucose <40
|
|
|
What is a normal ammonia level?
|
Normal < 50 µmol
|
|
|
What level of ammonia would make you worry about an inborn error of metabolism? What does this tell you depending on how old the infant is?
|
>200 µmol
- If within 24 hours or preterm → Transient Hyperammonemia of Newborn - If after 24 hours → inborn error of metabolism |
|
|
A patient with a metabolic acidosis with increased anion gap has a normal lactate and abnormal organic acids, what is this?
|
Organic Acidemia
|
|
|
A patient with a metabolic acidosis with increased anion gap has an elevated lactate and abnormal organic acids, what could cause this?
|
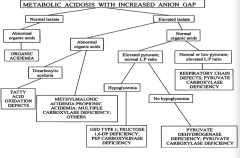
- Dicarboxylic aciduria = Fatty Acid Oxidation Defect
- Methylmalonic acidemia, propionic acidemia, multiple carboxylase deficiency, other |
|
|
A patient with a metabolic acidosis with increased anion gap has an elevated lactate and normal organic acids and elevated pyruvate with normal L:P ratio, what could cause this?
|
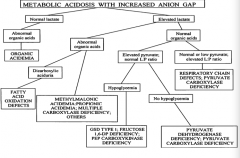
- Hypoglycemia → GSD type 1, fructose-1,6-DP deficiency, PEP carboxykinase deficiency
- No hypoglycemia → pyruvate dehydrogenase deficiency, pyruvate carboxylase deficiency |
|
|
A patient with a metabolic acidosis with increased anion gap has an elevated lactate and normal organic acids and normal or low pyruvate with elevated L:P ratio, what could cause this?
|
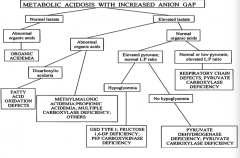
- Respiratory chain defects
- Pyruvate carboxylase deficiency |
|
|
If a neonate has symptoms of hyperammonemia within the first 24 hours of life and is preterm, what is the diagnosis?
|
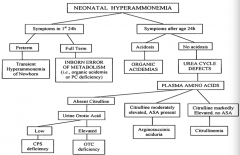
Transient Hyperammonemia of Newborn (THAN)
|
|
|
If a neonate has symptoms of hyperammonemia within the first 24 hours of life and is full term, what is the diagnosis?
|
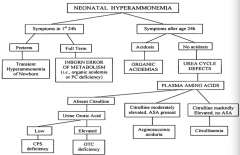
Inborn Error of Metabolism (ie, organic acidemia or PC deficiency)
|
|
|
If a neonate has symptoms of hyperammonemia after 24 hours of life and is acidotic, what is the diagnosis?
|
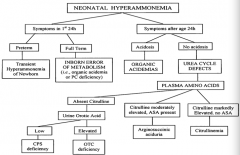
Organic Acidemia
|
|
|
If a neonate has symptoms of hyperammonemia after 24 hours of life and is not acidotic, what is the diagnosis?
|
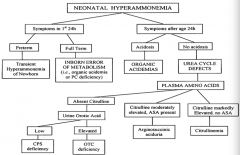
Urea Cycle Defect
|
|
|
If a neonate has symptoms of hyperammonemia after 24 hours of life and is not acidotic, with absent citrulline, what is the diagnosis?
|
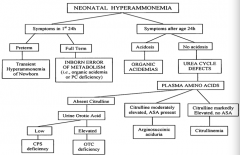
- If urine orotic acid is low → CPS deficiency
- If urine orotic acid is high → OTC deficiency |
|
|
If a neonate has symptoms of hyperammonemia after 24 hours of life and is not acidotic, with moderately elevated citrulline and ASA present, what is the diagnosis?
|
Arginosuccinic aciduria
|
|
|
If a neonate has symptoms of hyperammonemia after 24 hours of life and is not acidotic, with markedly elevated citrulline and no ASA present, what is the diagnosis?
|
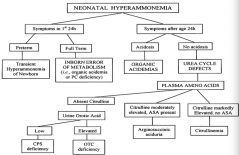
Citrullinemia
|
|
|
Case 1:
- 3 day old male is brought to ED w/ lethargy progressing to unresponsiveness - Baby was born full term, APGARs 8 and 9 at 1 and 5 minutes - Feeding normally for 24 hours but became irritable and less interested in feeding - Notice he is breathing fast and deep and is unresponsive - Labs: Na+ 136, K+ 4.8, Cl- 101, HCO3- 10, BUN 26, Cr 0.7, blood glucose 96 What is this infant's anion gap? |
25
Na+ - (Cl- + HCO3-) |
|
|
Where are organic acids metabolized?
|
Mitochondria
|
|
|
What happens if there is a block in the metabolism of organic acids?
|
Elevation of specific acylcarnitine, which are identified by the plasma acylcarnitine profile
|
|
|
If a newborn has ketonuria, what do you think?
|
Inborn error of metabolism
|
|
|
What are the signs of organic acidemia?
|
- Urine organic acids
- Ketonuria - Neutropenia, thrombocytopenia - +/- Hyperammonemia - Abnormal acylcarnitine on newborn state screen |
|
|
How do you treat a patient with organic acidemia?
|
- Stabilize
- Get rid of organic acid intermediates and ammonia → hemodialysis - Give carnitine - After stabilization, may resume oral feeds - Consult dietitian and metabolic specialist |
|
|
What would you give to all babies with organic acid aciduria?
a) Ornithine b) Carnitine c) Vitamin B12 (cobalamine) d) Biotin (Vitamin B7) e) Folate (Vitamin B9) |
Carnitine
|
|
|
Case 2:
- 3 day old male is brought to ED w/ history of lethargy progressing to unresponsiveness - Born full term, APGARs 8 & 9 at 1 and 5 min - Feeding normally for 24 hours but became irritable and less interested in feeding - He is breathing fast and deep and is unresponsive - Labs: Na+ 139, K+ 4.1, Cl- 101, HCO3- 26, BUN 20, Cr 0.5, blood glucose 96 What is this infant’s anion gap? |
12
Anion gap = Na+ - (Cl- + HCO3-) |
|
|
Case 2:
- 3 day old male is brought to ED w/ history of lethargy progressing to unresponsiveness - Born full term, APGARs 8 & 9 at 1 and 5 min - Feeding normally for 24 hours but became irritable and less interested in feeding - He is breathing fast and deep and is unresponsive - Labs: Na+ 139, K+ 4.1, Cl- 101, HCO3- 26, BUN 20, Cr 0.5, blood glucose 96 - Blood gas: pH 7.50, PaCO2 20, HCO3- 25, BE 1 - Urinalysis: no ketones - Ammonia >1400 What is wrong? |
- Hyperammonemia
- Respiratory alkalosis Inborn error in urea cycle |
|
|
What are the signs of a urea cycle disorder?
|
- No acidosis (respiratory alkalosis)
- No ketones (unlike organic acidemia) - No hypoglycemia - Significant hyperammonemia |
|
|
What would build up if there is a deficiency / defect in ornithine trasncarbamylase? How is this inherited?
|
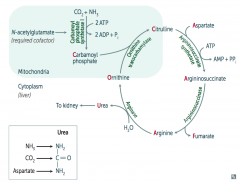
Build up of Ornithine and Carbamoyl Phosphate (X-linked)
|
|
|
What would build up if there is a deficiency / defect in argininosuccinate synthetase? How is this inherited?
|
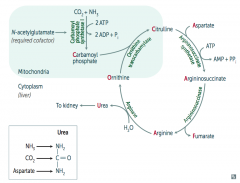
Citrulline (autosomal recessive)
|
|
|
What would build up if there is a deficiency / defect in argininosuccinate lyase? How is this inherited?
|
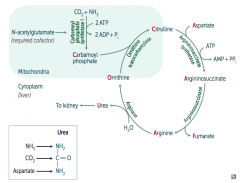
Argininosuccinate (autosomal recessive)
|
|
|
What would build up if there is a deficiency / defect in arginase? How is this inherited?
|
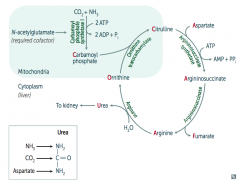
Arginine (autosomal recessive)
|
|
|
How do you treat children with urea cycle disorders?
|
- Hydration with D10 + electrolytes
- Discontinue all protein for 24 hours (calories must come from only carbs and fat) - Remove ammonia via hemodialysis and Na+ phenylacetate / Na+ benzoate - Give arginine - Protein restriction for life |
|
|
What lifetime changes must a patient with a urea cycle disorder make?
|
- Must restrict protein in diet for life
- Nitrogen waste medications - Aggressive treatment for illness - Liver transplant |
|
|
What is the prognosis for patients with a urea cycle disorder?
|
- Prognosis: guarded
- Even with treatment, many will die or develop significant development delays - Definitive treatment: liver transplant |
|
|
What enzyme is deficient / defective if you need to treat with arginine?
|
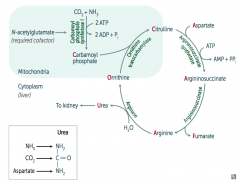
Argininosuccinate Lyase
|
|
|
What is the most common type of urea cycle deficiency?
|
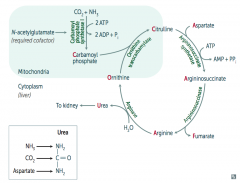
Ornithine Transcarbamylase deficiency (X-linked recessive)
|
|
|
What are the types of aminoacidopathies?
|
Phenylketonuria
|
|
|
What is deficient in Phenylketonuria? What builds up?
|
Deficiency in phenylalanine hydroxylase
- Elevated phenylalanine is neurotoxic |
|
|
How do you get Phenylketonuria?
|
Autosomal recessive
|
|
|
What are the consequences of untreated Phenylketonuria?
|
- Impairs neurological development leading to intellectual deficits (mental retardation, cognitive disability, intellectual disability)
- Fair colored skin / hair - Skin issues (eczema) - Musty body odor |
|
|
What are the consequences of Phenylketonuria that is treated with an appropriate diet?
|
Normal
|
|
|
How do you treat Phenylketonuria?
|
- Limit / restrict Phenylalanine
- Provide Tyrosine (because it is normally made from Phenylalanine by Phenylalanine Hydroxylase - which is deficient) |
|
|
What is involved in the emergency treatment of any inborn error of metabolism?
|
- ABCs: Airway Breathing Circulation
- Treat dehydration and infection - Treat seizures - Whole body cooling to prevent damage |
|
|
What are the first steps in metabolic therapy for inborn errors of metabolism?
|
- Reduce precursor substrate load
- Provide caloric support - Provide fluid support - Remove metabolites - Divert metabolites - Supplement with cofactors |
|
|
What are the characteristics of the newborn state screen?
|
- WI screens for 44 disorders by Tandem Mass Spectrometry (MS/MS)
- Heal stick at 36-48 hours of life - Filter paper sent to state lab within 24 hours - Results within 48 hours |
|
|
Treatment goals for IEM include which of the following aims?
a) Reduce precursor substrate load b) Provide caloric and fluid support c) Remove metabolites d) Divert metabolites with supplement with cofactor(s) e) All of the above |
All of the above
|
|
|
What are the characteristics of Newborn State Screening Programs?
a) Have no risks b) Prevent developmental disabilities c) Are an added cost to health care expenditure d) Are mandatory for all infants e) All of the above |
Prevent Developmental Disabilities
|
|
|
What is the only absolute sign of ovulation?
a) Regular menstrual cycles b) Increased cervical mucus at mid-cycle c) Pregnancy d) Mittelschmerz e) None of above |
Pregnancy
|
|
|
When is intra-cytoplasmic sperm injection recommended?
|
• Very low sperm count
• Poor morphology or motility • Failed IVF attempts • Post-vasectomy collection from epididymis or testicle • ↓ Erection/ejaculation: spinal cord injury, diabetes, etc. |
|
|
When are the risks of intra-cytoplasmic sperm injection?
|
• Risk genetic & developmental defects, but underlying problem at play
• Male child inherit father’s infertility • ↑ miscarriage • Genetic problems passed to male child, e.g. cystic fibrosis |
|
|
What are the early pregnancy risks of maternal death?
|
- Ectopic pregnancy
- Choriocarcinoma |
|
|
A 32 year old sexually active female is 8 weeks from last menses. She presents to ER with complaints consistent with appendicitis. A pregnancy test is pending. What is the most likely diagnosis?
|
Ectopic Pregnancy
|
|
|
Where do most ectopic pregnancies occur? Where else?
|
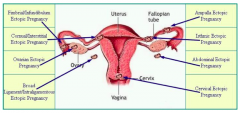
- 95% occur in some portion of the fallopian tube
- 5% occur in other locations |
|
|
What is necessary for evaluating an ectopic pregnancy?
|
- Cannot rely purely on physical exam
- Ultrasound exam should be part of the initial evaluation - If transvaginal ultrasound does not detect intrauterine pregnancy, presumptive ectopic pregnancy is virtually certain when the serum β-hCG level is >1500/mL |
|
|
What needs to happen if there is a ruptured ectopic pregnancy?
|
Warrants surgery
|
|
|
What needs to happen if there is the ectopic pregnancy is not ruptured?
|
Warrants observation if hCG is waning, otherwise methotrexate therapy
|
|
|
What genes have more control over placental growth?
|
Paternal genes
|
|
|
What genes have more control over fetal growth?
|
Maternal genes
|
|
|
What are the types of Gestational Trophoblastic Disease?
|
- Hydatidiform mole
- Choriocarcinoma |
|
|
What happens in Gestational Trophoblastic Disease?
|
A sperm cell fertilizes an abnormal egg cell that has no nucleus (or chromosome) and divides OR 2 sperm fertilize an empty egg cell
|
|
|
How do you treat a hydatidiform mole?
|
- Follow serum hCG levels (10^4 to 10^5)
- D&C Procedure - Follow hCG levels until it gets to 0 |
|
|
What can a choriocarcinoma spread to?
|
- Lung
- Spleen - Kidney - GI - Liver - Brain |
|
|
How do you treat a patient with choriocarcinoma?
|
- Anti-folate chemotherapeutic agent: methotrexate
- Follow hCG to 0 |
|
|
Which of the following chemotherapeutic agents is a folic acid antagonist?
a) 6-mercatopurine b) vincristine c) methotrexate d) paclitaxel e) cisplatin |
Methotrexate
|
|
|
Velamentous umbilical cord insertions are associated with:
a) More congenital anomalies than with central cord insertions b) Fetal distress/death if vasa previa c) Retained placenta d) Higher rate of spontaneous abortion e) All of the above |
All of the above
|
|
|
What is a Velamentous umbilial cord?
|
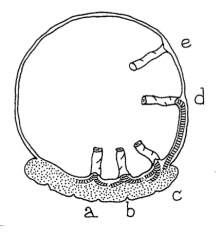
- Umbilical cord inserts into the fetal membranes (choriamniotic membranes), then travels within the membranes to the placenta (between the amnion and the chorion)
- The exposed vessels are not protected by Wharton's jelly and hence are vulnerable to rupture |
|
|
What are the types of twins?
|
- Dizygous (multiple ova, fraternal, familial, 2/3)
- Monozygous (single ovum, not thought familial, 1/3) |
|
|
What are the types of placentas based on the type of twins?
|
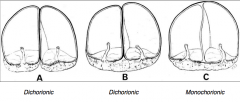
- Dizygous twins always have a separate placental disc (Dichorionic)
- Monozygous twins may have separate or shared placentas (Dichorionic or Monochorionic) |
|
|
What are the risks of monozygotic twins?
|
- More likely to be born premature
- More likely to be growth discordant - More likely to have developmental anomalies - More likely to have twin mortality |
|
|
What usually accounts for the excess monozygotic morbidity and mortality?
|
Almost always monozygotic twins with MONOCHORIONIC placentas
|
|
|
Which multiple gestation is the most high risk?
a) Dizygotic dichorionic b) Monozygotic monochorionic c) Monozygotic dichorionic d) Monozygotic trichorionic |
Monozygotic Monochorionic
|
|
|
When is there increased risk of small intestinal atresia?
|
Twins
- Especially among same-sex twins - Jejunoileal > ileal atresia suggests vascular cause - Possible disruption in Monozygotic Monochorionic twins |