Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
18 Cards in this Set
- Front
- Back
|
Write down the five sub-parts of the formula that every molecular dynamics program seems to have included in the source code.
|
5 bonded and non-bonded interactions
|
|
|
Why should we use structures solved at 1.2 Ångström or better?
|
We use A-ray structures at the highest possible resolution. 1.2 Ångström resolution is a nice compromise. Also 1.2 Ångström is often called the limit of atomic resolution.
|
|
|
Think of a few very different ways to arrive at R0 values.
|
Quantum chemistry; XAFS; The method we are doing now but applied to the CCD; etc.
|
|
|
Summarize the algorithm we chose here, in this experiment.
|
We look at a distribution and use the inverse Boltzmann law. The concept is that if we see
something more often, it must be more favourable. |
|
|
How would you design the software for the elevator in the Radboud parking house?
Why is this a much more difficult task than makeing a force field to predict the secondary structure of proteins? |
1) Collect data about the times that people push the 'elevator come here button' and register where they go once the elevator is here.
2) Use this to determine the time-dependent most likely next floor where somebody will push the button, and send the elevator there when it has nothing to do. 3) But I admit that is energetically not optimal, and most complications are overseen. For example, in the morning, the garage fills up from the bottom up, in the afternoon it empties much less regularly. This can imply that the morning software will differ from the afternoon software. In the afternoon people enter at the ground floor in the morning every 10 minutes mostly one floor higher... 4) Other complications are that things differ when it rains, when the vier-daagse is ongoing, when it is spring vacation, etc. |
|
|
How would you write software to predict if a residue is an active site residue or not?
Don't forget that active sites are always at the surface of the protein. Think of the data-set you need. Think of the null-model. Think of the real model, and how to convert counting into scoring. Think about testing the method. |
The data-set needed is a large set of proteins with known active sites.
You then have to determine how often each residue type occurs at the surface in the whole set. Suppose you have 7.2% Ala, 1.3% Cys, 5.5% Asp, etc), then this is your null-model. If you now find a protein you expect any local area to have roughly this same distribution. Now you count the residue types in the active site pockets. Of course, it requires a bit of thinking where the active site stops and the 'rest' begins, but that is a matter of definition. The number of active site residues is going to be much lower than the number of surface residues, so these two rows of 20 numbers must be scaled on each other. After that, you can divide the two rows of 20 numbers on each other, and take the logarithm, and those logaritms are your scores. Now, if you have a protein with a nice dent in its surface, you can count the residue types present at the surface of this dent, and perhaps also count the homologous positions in a couple dozen homologs to get better statistics. But however you count, this score is related to the likelyhood that your protein's dent is not just a dent, but an active site. |
|
|
If you call things wrong when they deviate more than 3σ from the theoretical mean, how big is then the risk that you call something wrong that is actually right?
|
Wikipedia tells me:
1σ 68.3% 2σ 95.4% 3σ 99.7% 4σ 99.994% 5σ 99.99994% 6σ 99.9999998% 7σ 99.9999999997% Wikipedia. So if I call everything wrong that deviates more than 3σ from the (real) mean, I take a 0.3% chance that I call something wrong that is right. |
|
|
What is a Z-score?
|
The number of σs that an observation deviates from the (real) mean. So if some value is 3.2σ too big, we call its Z-score=3.2.
|
|
|
Most checks compare aspects of one structure with the average of a series of well-solved structures. But for bond-lengths and bond-angles the PDBREPORT seems to have better values. How come we know the perfect bond-length?
|
Because it is determined from data in the CSD (Cambridge small molecule database) and those data are muuuuch more accurate and precise than the data in the PDB.
|
|
|
Which checks are useless for homology models?
|
For example crystal packing checks, or anything else related to crystals.
|
|
|
What is packing quality ?
|
The normality of packing as determined from a statistical comparison between the packing in the protein structure to be checked and the average packing of all proteins in a database of high resolution / high quality proteins.
|
|
|
What are side-chain flips? And how come we find so many of those in the PDB?
|
When in Gln or Asn the N and O in the side chain sit 'the wrong way around', or when the whole ring in His must be rotated 180o around the Cβ-Cγ bond. This is mainly cause by the fact that crystallographers normally cannot see the difference between N, C and O.
|
|
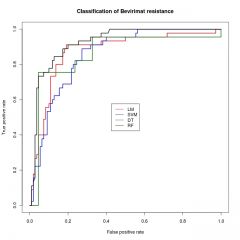
Explain this figure. What is on the axes? (I can read, so don't copy the legends, but explain them). And what do LM, SVM, DT, and RF actually stand for?
ROC curve for four R classifiers |
Random Forests etc
|
|
|
What is an EF-hand? And why do we use EF-hand motifs in this study?
|
EF hand is special kind of calcium binding loop (see wikipedia). And we use this because it can bind calcium, a major component of bone, hoping to use the empty EF-hand to bind calcium in bone.
|
|
|
Make the ROC curve for the prediction of calcium binding loops.
|
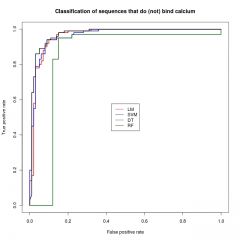
|
|
|
What is the major problem when using force fields?
|
Finding the correct null-model
Takes very much CPU time to do it right Sometimes we lack data to properly design the force field |
|
|
Mention a few terms (formulas not really needed) that are used in the force field of a molecular dynamics software package. And mention a few terms that generally are not yet in use in such force field?
|
In:
Bond lengths, angles and torsion angles VdW and charge-charge interactions, sometimes also H-H bonds and planarities Out: pi-pi stacking, induced polarities, quantum related fx |
|
|
What is a ROC curve?
|
receiver operating characteristic
sensitivity (true positives) plotted against specificity (false positives) |