Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
61 Cards in this Set
- Front
- Back

|
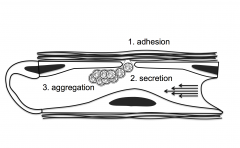
This is a platelet. Important for 1' hemostasis. Anucleate cells. Contain granules. Two types of vesicles. Vesicle contains an electron-dense core. Contractile proteins. Membranes - internal membrane system. Surface canalicular system.
|
|
|
Platelets do three things when there is damage to the endothelium: adhere, then become activated and undergo secretion (which attracts additional platelets) followed by aggregation (which results in a platelet plug leading to primary hemostasis).
|
|
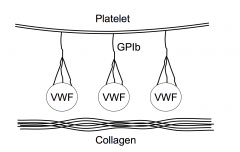
Platelet adhesion
|
vWF plasma protein mediates the interaction of the platelet with collagen. GP1b not seen on any other cell -- gives the platelet the unique ability to bind to VWF which lets it bind to collagen.
|
|
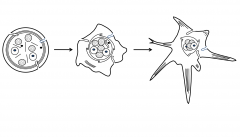
Once a platelet adheres or encounters a soluble agonist
|
get a fairly profound set of biochemical signals that lead to changes in platelet shape. Pseudopods stick out and can initiate the interaction with collagen or the interaction with other platelets. While changes in platelet shape are occurring -- there are contractile events occurring due to increased calcium. There is fusion of the cannalicular system and content of granules are dumped to the outside of the cell.
|
|
|
Platelet storage granules
|
1. Dense (Delta) Granules
ADP, ATP (agonist for platelet aggregation) Serotonin Calcium 2. Alpha Granules Fibrinogen, Factor V Thrombospondin PDGF, PF4, bTG 3. Lysosomal (Lambda) Granules |
|

|
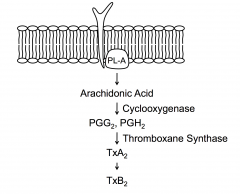
Membrane with agonist receptor in membrane.
In platelets one of the primary events - PL-A cleaves phospholipids generating arachidonic acid. Arachidonic acid can then be converted to PGG2 as shown etc Mechanism thought to help membranes fuse. |
|
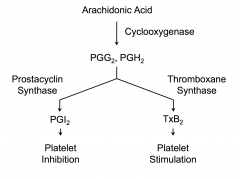
COX present in platelets and endothelial cells
|
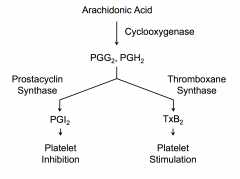
Without prostacyclin, you'd be forming platelet aggregates all the time and that wouldn't be good.
|
|
|
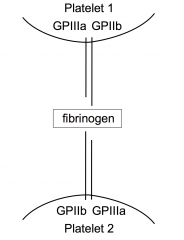
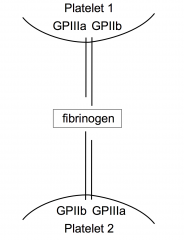
Integrins on platelet surface mediate platelet interaction
GPIIb interacts with fibrinogen which interacts with GPIIb and GPIIIa resulting in platelet aggregation |
|
|
Platelet Disorders
|
Quantitative:
Decreased platelet count Prolonged bleeding time Qualitative: Normal platelet count Prolonged bleeding time |
|
|
Defects of platelet adhesion
|
Cofactor Abnormal:
- Von Willebrand's Disease Platelet Abnormal: - Bernard-Soulier Syndrome Substrate Abnormal (collagen): Ehlers Danlos Syndrome Scurvy Pseudoxanthoma Elasticum |
|
|
Defects of Secretion
|
Prostaglandin Defects:
- Phospholipase defect - Cyclooxygenase defect - ASA and NSAIDs - Thromboxane synthase defect Storage pool defect - Alpha-SPD - Delta-SPD - Alpha-Delta-SPD |
|
|
Defects of aggregation
|
Membrane changes:
- Glanzmann's thrombasthenia Abnormal Mileau - Uremia - Fibrin split products |
|
|
Defects with Increased Destruction
|
1. Increased utilization
-- Diffuse intravascular coagulation -- Thrombotic thrombocytopenia purpura -- Abnormal hear valves 2. Immunologic Destruction -- Idiopathic thrombocytopenia purpura -- Lupus erythematosus Drugs - Quinidine |
|
|
Classification of ITP (idiopathic thrombocytopenia purpura)
|
1. Acute ITP
-- prominent bleeding and purpura -- frequently preceded by a viral syndrome -- No prior bleeding history -- usually children (M:F ~ 1:1) 2. Chronic ITP -- bleeding less prominent -- long history of easy bruising -- Usually affects adults (F>M) -- Indolent |
|
|
Characteristics of ITP antibodies
|
1. Platelet specific
2. Restricted heterogeneity (IgG3 - kappa and lambda) 3. Antigenic heterogeniety (GPIb, GPIIb, GPIIIa) 4. Site of production - spleen, bone marrow |
|
|
Diagnosis of ITP
|
1. Evidence of immune thrombocytolysis
-- thrombocytopenia -- normal or increased megakaryocytes -- anti-platelet antibody 2. Absence of other causes of thrombocytopenia -- lupus erythematosus -- diffuse intravascular coagulation -- drugs -- lymphoma |
|
|
Therapy of ITP
|
1. Corticosteroids (1-2 mg/kg/day)
-- 70% response with high relapse rate -- decreases antigen processing and antibody production 2. Splenectomy -- 50% response with low relapse rate -- removes antibody source and site of sequestration -- pneumovax in children 3. Immunosuppressive agents |
|
|
Most appropriate study for patient with issues with bleeding -- leading to bruising
|
Peripheral blood smear
|
|
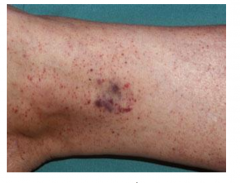
Differential diagnosis for patient with thrombocytopenia
|
Immune thrombocytopenia (ITP)
-- drugs -- infections -- autoimmune syndromes -- DIC -- TTP -- hematologic malignancy |
|

|
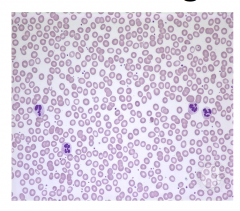
Patient is diagnoses with ITP. Platelets are fairly large. Almost same size of RBCs. The platelets have granules.
Treatment is with prednisone and IVIg is initiated and her platelet count responds to normal levels. Petechiae resolves. |
|
|
What would confirm Iron Deficiency Anemia?
|
-- Ferritin
-- Iron (transferrin) saturation -- Total iron binding capacity - Reticulocyte count |
|

|

|
|
|
Aspirin Effects
|
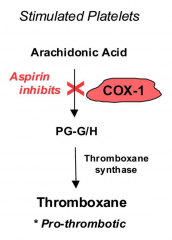
- Aspirin decreases platelet aggregation by inhibition in COX-1
- Results in decreased thromboxane formation - Thromboxane regulates platelet aggregation through mediating glycoprotein IIb/IIIa |
|
|
Defects of primary hemostasis (formation of platelet plug) can cause?
|
Mucocutaneous bleeding including petechiae and mucosal bleeding (epistaxis, gingival bleeding, menorrhagia)
|
|
|
Disorders that affect primary hemostasis include
|
Qualitative and quantitative platelet defects, vW disease, and disorders of the endothelium
|
|
|
ITP diagnosis
|
Diagnosis of exclusion requiring lab testing to evaluate for other causes of bleeding
|
|

|
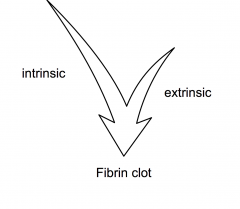
Most coagulation - extrinsic
intrinsic - link to inflammatory system and amplification |
|

|

|
|

|

|
|

|
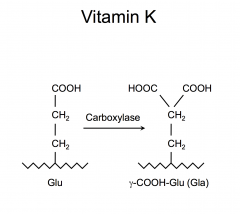
gamma-carboxy allows for binding of calcium with is important conformational changes in proteins - necessary for their interaction with phospholipid surfaces.
|
|

|
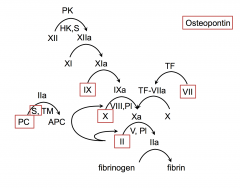
vit K dependent proteins ; factors 2,7,9 and 10
|
|
|
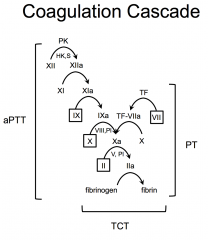
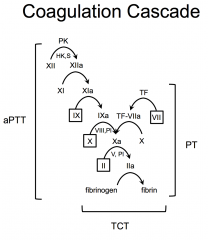
aPTT assesses intrinsic coagulation
PT assesses extrinsic coagulation TCT assess the ability of fibrinogen to convert to fibrin |
|
|
Isolated Prolonged aPTT
|
High Molecular Weight Kininogen deficiency
Prekallikrein deficiency FXII deficiency FXI deficiency FIX deficiency FVIII deficiency Passovoy Deficiency Lupus anticoagulant |
|

|
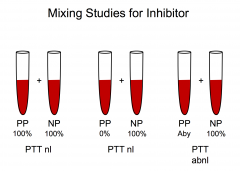
Mixing study: take a patients plasma and normal plasma and mix them together.
|
|
|
Isolated Prolonged PT
|
FVII deficiency
|
|
|
Prolonged PT and aPTT
|
FX deficiency
FV deficiency FII deficiency Fibrinogen deficiency |
|
|
Isolated Prolonged TCT
|
Afibrinogenemia
Hypofibrinogenemia Hyperfibrinogenemia Dysfibrinogenemia FDPs Immunogloblins Heparin |
|
|
Bleeding with Normal Screens
|
Mild factor deficiency
Thrombocytopenia Thrombocytopathia Vascular abnormality (scurvy) FXIII deficiency Antiplasmin deficiency |
|
|
Test for post-operative bleeding
|
Partial thromboplastintime 1:1 mixing study
|
|
|
Management for a patient with post-operative bleeding
|
- Assess patient's airway with intubation if required
- IV fluids - Type and cross blood for transfusion - Surgical hemostasis - Further lab studies |
|
PTT (repeat)= 42 seconds (nml 25-32)
PTT 1:1 mix = 31 seconds Von Willebrand’s antigen= 88% (nml 60-130) Von Willebrand’s activity = 115% (nml 50-130) What laboratory result would best explain the patient's bleeding complications |
Decreased factor VIII activity
-- has mild hemophilia A |
|
|
Hemophilia
|
An inherited bleeding disorder in which there is a deficiency or lack of factor VIII (hemophilia A) or factor IX (hemophilia B)
|
|
|
Inheritance of Hemophilia
|
Hemophilia A and B are X-linked recessive disorders
Hemophilia is typically expressed in males and carried by females Severity level is consistent between family members |
|
|
Degrees of Severity of Hemophilia
|
Mild Hemophilia:
factor VIII or IX level = 6-50% Moderate Hemophilia factor VIII or IX level = 1-5% Severe Hemophilia factor VIII or IX level = < 1% |
|
|
Pattern or bleeding most commonly identified in hemophilia?
|
Joint bleeding - hemarthrosis
Muscle hemorrhage Soft tissue Life threatening-bleeding --- intracranial, neck, ileopsoas, or retroperitoneal Other - surgery |
|
|
Development of an inhibitor (antibody) against factor VIII
|
Polyclonal high affinity IgG Ab that neutralize the procoagulant function of FVIII or FIX
Factor replacement therapy becomes ineffective unless the antibody is removed Most serious complication of factor replacement therapy Presents major economic and clinical challenges |
|
|
A 44-year-old woman presents for her annual exam. Findings at the physical exam are BP-120/80 mmHg, HR - 70 beats/min, pale mucous membranes, and increased uterine size. The patient also presents with
menorrhagia of 2 years duration. Her blood test shows microcytic hypochromic red blood cells (RBCs). Further exams show low serum iron, increased total iron-binding capacity (TIBC), and low ferritin. What is the most appropriate diagnosis for this patient? |
Anemia secondary to iron deficiency
|
|
|
A 14-year-old female is brought to the Family Medicine Clinic presenting with heavy menses and
occasional epistaxis. The mother reports that this is common among the women in their extended family but not investigated until now. Lab values in the preliminary workup include a set of abnormal closure times in the PFA-100 bleeding test device (but not indicative of aspirin effect), a normal PT, slightly prolonged aPTT, normal platelet count, normal fibrinogen, and Hematocrit=32%. A von Willebrands workup reveals 30% antigen level, 25% ristocetin cofactor activity level, and 40% Factor VIII clotting activity. What is the most likely diagnosis to explain the bleeding in this patient? |
Congenital von Willebrands
disease Type I |
|
|
An 18-year-old woman needs to be
scheduled for an elective cholecystectomy and was assessed to have a possible bleeding disorder. She reports frequent bruising, episodic epistaxis, bleeding gums, and heavy menstrual cycles for a few years. She takes no medications and has no medical illnesses. What lab results will support a diagnosis of von Willebrand’s disease in this patient? |
Prolonged partial thromboplastin and
bleeding times |
|
|
A mother comes to the pediatrician with her
5-year-old daughter. She complains that her daughter’s gums bleed when she bushes her teeth. The daughter’s nose also bleeds 2 or 3 times a month. At the physical exam, there are no important findings. Lab tests report that platelets do not aggregate in response to ristocetin and the defect corrects with addition of normal plasma. |
Von Willebrand syndrome
|
|
|
10 children who attend the state fair as a group fall
ill the next day with bloody diarrhea. As a primary care physician, you are managing 3 of these patients with fluids and an antimotility drug. On the 4th day of your care, fever and symptoms have abated in one patient but the other two show decreasing urinary output. Labs reveal, in both patients, rising creatinine levels and falling red cell and platelet counts, while PT and aPTT are normal. The blood smears reveal microangiopathic changes with schistocytes. Direct Coombs test is negative. The symptoms progress rapidly and become more severe. Frank blood continues in each stool, purpura and petechiae are present, the patients are anuric and hypertensive, and hemolysis is visually evident in blood plasma samples. What would be the most beneficial treatment modality for these patients at this point? |
Daily plasma exchange
|
|

What is a characteristic of the disorder that you would expect to find?
|
Anti-platelet IgG
|
|

After a few months, which of the
following would you expect to see in this patient's peripheral blood smears? |
Howell-Jolly bodies
|
|

What is the most prudent course of
action? |
Order a workup of contact activation factors, but proceed with the angioplasty
|
|

What special diagnostic test will indicate
and confirm the etiology of the disease? |
ADAMTS-13 metalloprotease antigen and activity test for cleavage of multimeric von Willebrands factor
|
|

What is the most probable diagnosis
for this child? |
beta-thalassemia
|
|

What is the most likely etiology of
hemolytic disease in this patient? |
Exposure to particular E.Coli strain at the fair, producing toxic endothelial damage and microthrombi
|
|
|
A 42-year-old African American man sustains severe
injuries in an automobile accident and is admitted to the intensive care unit. Examination of a peripheral blood smear on the 3rd day of admission reveals helmet cells, schistocytes, and decreased platelets. What condition is most strongly suggested by these findings? |
DIC
|
|

Given his history, what should the
physician do in addition to monitoring prothrombin time? |
Decrease dosage of warfarin
|
|
|
A 21-year-old male is known to have hemophilia B. He
has frequent bleeding into his large joints. Recently, he was hospitalized for a GI bleed. Which of the following laboratory test results would most likely be abnormal secondary to the underlying bleeding disorder? |
Activated partial thromboplastin time
|
|
|
A 4-year-old boy begins to develop hematomas. He
complains to his mother of knee pain, and she notices they are swollen and painful. His mother is very concerned because she knows her father had a bleeding disorder. It is determined that this boy has hemophilia A because he is deficient in what factor? |
VIII
|