Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
217 Cards in this Set
- Front
- Back
|
Kidney cortex
|
- contains the glomeruli (juxtamedullary and cortical) and the lowest glomerulus defines the end of the cortex (or the corticomedullary border).
- contains most of the kidney’s vasculature and so receives about 75% of the blood flow to the kidney. - The interstitial space is isosmotic with plasma. |
|
|
Kidney medulla
|
- seperated into the inner medulla (deeper) and outer medulla (next to cortex, contains the thick loop of Henle)
- contains the loop of Henle and the descending osmolarity gradient in the intersitial space (also has the vasa recta which maintains the gradient) |
|
|
Renal Pelvis
|
- connects the kidneys to the ureters
- nephrons drain filtrate into the collecting ducts which connect to the minor calyxes and the the major calyx and the renal pelvis. |
|
|
Functions of the glomerulus
|
= a bundle of capillaries fed by an afferent arteriole (drained by and efferent arteriole), sitting in the Bowman's capsule.
- first filtration step in the kidney: Filters based on size (also charge), allows small molecules (ions, glucose, water) to be pulled out of the plasma into the Bowman's space (RBC's and proteins do not pass) - Filters an average of 180L/day of blood |
|
|
Functions of the proximal tubule
|
- connects the glomerulus to the loop of Henley (located in the cortex). Has a brush border for max surface area, associated with the peritubular capillaries
- responsible for the bulk of material reabsorption: 65% Na & Cl (but doesn't change [Na]), 65% H2O, 50% of K, Ca, & urea, 90% HCO3, 100% organic nutrients (glucose) - will secrete foreign substances and drugs |
|
|
Functions of the loop of Henle
|
- located in the medulla, recieves isoosmotic fluid from the proximal tubule and delivers hypoosmotic fluid to the distal convoluted tubule, associated with the vasa recta (together establish interstitial gradient)
- Water escapes in the thin descending limb from aquaporins - Thick ascending limb is impermeable to water but allows diffusion/pumping of Na - Main transporter of Na in the ascending limb is NKCC (moves Na, K, 2Cl in on apical side), Na/K exchanges on basal side (K, Cl diffuse through channels) |
|
|
Functions of the kidney
|
1. Regulation of water and electrolyte balance.
2. Excretion of metabolic waste. 3. Excretion of foreign substances. 4. Regulation of ECF volume (and indirectly blood volume and pressure). 5. Regulation of red blood cell production. 6. Regulation of the active form of Vit. D. 7. Regulation of acid/base balance. |
|
|
Salt and water reabsorption in the proximal tubule
|
Step 1 - Passive apical entry of Na+. Active Na+ exit at basolateral membrane (recycle K+ at basolateral membrane). Movement of cations establishes lumen negative potential.
Step 2— Potential drives movement of anions from lumen to interstitium and establishes an osmotic gradient. Step 3- Osmotic gradient drives water movement from lumen to interstitium. Step 4— Accumulation of water and salt in interstitium and Starling’s forces promotes bulk flow of water and salt into the peritubular capillaries. |
|
|
Functions of the distal convoluted tubule
|
- Receives slightly hypoosmotic filtrate from the loop of Henle, delivers it to conducting duct
- Na/Cl symporter pumps small amounts of Na & Cl out of the filtrate, further diluting filtrate |
|
|
Functions of the collecting duct
|
Cortical collecting duct: receives filtrate from DCT
- reabsorbes some Na+ (ENaC channel) and H2O, secretes K+ Medullary collecting duct - involved in the secretion of acids and bases, reabsorption of water and urea (based on hormone stimulation). - Delivers fluid for excretion to the calyxes |
|
|
Components of the glomerular filtration apparatus
|
- Endothelial cells: line the capillaries in the in glomerulus. They have fenestrated pores that selectively filter plasma (~70nm)
- Basement Membrane: spongey, mesh of proteins secreted by endo & epithelial cells. Has a slight negative charge that confers minimal fitration based on charge - Podocytes: specialized epithelial cells (sit on basement membrane) with large nuclei and foot processes which interdigiate to form slit diaphragms which filter according to size. |
|
|
Factors affecting the net filtration pressure in the glomerulus
|
Pressure in:
- main force is the hydrostatic pressure of the afferent capillary (~60mmHg) Opposing pressures: (net ~45mmHg) - plasma oncotic: proteins cannot filter, so become more concentrated and pull water back into the plasma (30mmHg) - capsule hydrostatic pressure: pressure from fluid in the Bowman's capsule forces fluid back into capillaries (15mmHg) Net filtration pressure = capillary hydrostatic pressure - (capillary osmotic + capsular hydrostatic pressures) NFP = 15mmHg |
|
|
Glomerular filtration rate
|
GRF = amount of fluid filtered by glomerulus per unit time, proportional to net filtration pressure; GFR = K*NFP (where K is glomerular capillary filtration coefficient)
- maintained relatively constant over a wide range of blood pressures - overall GFR is 125mL/min or 180L/day in a normal adult. This decreases with age and renal disease |
|
|
Regulation of the GFR
|
Regulation mostly concerned w/ resistance of afferent arteriole (which matches pressure changes). Goal is to keep pressure almost constant
3 Mechanisms: 1. Autoregulation: intrinsic to the kidney; mechanical stretch in the arterial smooth muscle causes secretion of signaling molecules that change SM tone and lower resistance 2. Autonomic regulation: sympathetic neurons innervating the arteriole induce SM contraction during severe BP changes (vasoconstriction reduces GFR, keeping fluid in circulation). 3. Tubuloglomerular feedback: the macula densa (part of the TAL of Henle) sits in between the arteries and senses salt/water balance. Changes in salt in the distal nephron stimulates changes in blood flow or mesangial cell size in the glomerulus |
|
|
Renal Clearance
|
= removal of a substance from the blood and excretion in urine; excretion rate; volume per unit time.
- helps measure GFR experimentally: if a substance is freely filtered, not secreted or reabsorbed then filtration rate = excretion rate; GFR = urine flow rate x [sub](urine)/[sub]plasma - inulin used in labs, creatinine used in humans (gives over estimate of GFR b/c of secretion) - Most solutes have clearance < GFR b/c not freely filtered or lots of reabsorption: glucose clearance = 0, Na ~0.01GFR, Urea ~0.5GFR |
|
|
How to secrete dilute urine
|
Goal: excrete more salt and water. Max output: 20L/day, min [urine] = 50mOsm/L. Total amount of solute remains fairly constant
- Glom: little change, unless BP has gone up dramatically (>200mmHg) -Prox T: little change, reabsorb water and solutes, isoosmolar - LoH: Little change, H20 & Na reabsorb based on osmol differences DCT & CD: Most change: will simply let fluid go with little reabsorption. ADH will be low, so DCT impermable to H2O (can still reabsorb solutes). Also interstitial medulla gradient will be weaker (dilute filtrate) so less gradient to filter in CD. |
|
|
How to secrete concentrated urine
|
Goal: retain salt and H2O. Min output: 0.5L/day, Max [urine]= 1200-1400mOsm/L (4-5x plasma osmolarity)
Mediated by 3 factors: 1. high ADH (vasopressin): increases DCT and CD permeability by moving AQP2 channels to the luminal membrane 2. High osmolarity of the medullar interstitial fluid: provides gradient to move H2O in the presence of ADH - Medulla interstitium is concentrated by: active transport of Na/K/Cl out of TALoH creating gradient by counter current multiplication (distributed and maintained by vasa recta; counter current exchange); active transport of ions out of CDs; facilitated diffusion of urea from the CDs; minimal H2O diffusion into the medulla |
|
|
Counter current exchange & multiplication
|
- exchange of osmolarity between the tubes of the LoH because they run antiparallel and have different permeabilities
- Na/K pumps in the TAL dilute the filrate, and induce H2O diffusion from the TDL, concentrating it. As the fluid moves through the loop, these effects are multiplied, creating the gradient. (the longer the loop the greater the concentration) - the vasa recta also uses these mechanism to absorb and redistribute Na, maintaining the gradient (no active transporters), has slow flow allowing equilibration at different levels (fast flow would dissipate gradient) |
|
|
Urea recycling
|
- urea is pumped out of the lower collecting duct by the uniporter (impermeable at the top), concentrated in the medulla, and reabsorbed at the bottom of the loop of Henle.
- Contributes 50% of the medulla concentration gradient (allows salt to be excreted) |
|
|
ADH action in the nephron
|
- ADH = antidiuretic hormone/vasopressin.
- pituitary peptide hormone, secretion stimulated by high plasma osmolality (mainly) or by very low blood volume/pressure - acts on principle cells of the cortical and medullary collecting ducts to upregulate water reabsorption by triggering the fusion of AQP-2 containing vesicles with the luminal membrane (ADH receptor binding activates cAMP, PKA which P's AQP2 initiating transport) -ADH also enhances hyperosmotic medullary interstitium by increasing urea reabsorption (acts on transporters in CD) and Na reabsorption in CDs/TAL |
|
|
Potassium movement in the nephron
|
- K needs to be maintained in a narrow range because [ECF] is so low, even small changes can have a big effect (especially on cells that use electric gradients)
- K is freely filtered in the glom. 65% is reasorbed passively in the Prox T, 25% in the TAL, and 5-10% in the CD - in case of low [K], transporters int the CD are upregulated to reabsorb more - for high [K], K is secreted into the DCT (stimulated by high flow or aldosterone), and CD transporters are minimized. Can excreted up to 100% |
|
|
Calcium movement in the nephron
|
- Ca is controlled hormonally outside the kidney PTH (breaks down bone) and calcitriol (stimulates GI absorption, mediates bone formation)
- Kidneys produce calcitriol (active form of Vit. D after D2 intermediate) via PTH stimulation and excretes phosphate (prevents over secretion of PTH) - Most Ca in blood is bound/complexed - free Ca is reabsorbed passively in the Prox T (60%) ane LoH (30%). Active Na/Ca antitransporters in the DCT are stimulated by PTH (absorb 5-10% more) - normally <1% is excreted, if levels are too high a large Na load facilitates excretion b/c ions are coupled in the DCT |
|
|
Phosphate movement in the nephron
|
Regulated by 2 mechanisms:
- conversion to calcitriol (with Ca) or break down from it in bone - renal modulations: --5-10% is protein bound, of free 75% get actively reabsorbed in the Prox T via Na sympoter (Tm limited system) --Normal filtered load is higher than Tm (saturated) so the rest is excreted. PTH inhibits this reabsorption ---will act as a buffer for H+ |
|
|
Glucose movement in the nephron
|
- Gets freely filtered in the glom
- 100% gets reabsorbed in the Prox T via Na-Glu symporters (SGLT) on the luminal membrane. Then diffuses out via GLUT channels into the interstitium - no normal regulation, but in high load (diabetes) the transport can be overwhelmed and glucose will be excreted |
|
|
Renal response to high volume/pressure
|
- High BP/volume is sensed at the afferent arteriole as an increase in pressure across the golmerulus:
↑NFP → ↑GFR → ↑Filtrate load → Prox T. can't reabsorb as efficiently → ↓ Na reabsorption - In the distal nephron: ↑ Na (osmotic + weaker gradient) + ↑ Filtrate load + ↓ ADH (pituitary) → ↓ H2O/Na reabsorption + ↑ urine volume - Overall response: pressure naturesis (excess Na excretion) and pressure diuresis (excess H2O excretion) lowing blood volume and BP |
|
|
3 mechanisms that regulate Renin release
|
1. Sympathetic nervous stimulation: low pressure in arterial & cardiopulmonary baroreceptors causes CNS to stimulate renal nerves, releasing NE which acts on β1 adregenic receptors on granular cells (around afferent ateriole) which secrete renin
2. intrarenal baroreceptor: granular cells sense mechanical changes in flow, secrete renin if pressure is low 3. Salt & flow sensors: macula densa (btwn arterioles & glom) senses tubular flow rate & [salt]: - ↑GFR → ↑Flow/Na → detected by MD cilia in TAL → MD secretes signals that inhibit renin release & constrict afferent arteriole - ↑GFR → ↓Na/flow → MD senses ↓Na at TAL → stimulates renin release by granular cells & relax afferent arteriole (so bad waste still excreted) |
|
|
Renin & Angiotensin
|
Renin = enzyme that converts angiotensinogen (made in liver) to angiotensin I (then converted to active angiontensin II by angiontensin converting enzyme, ACE)
- Angiotensin then acts to conserve Na+ (therefore H2O) and increase BP 1. stimulates Prox T to increase Na reabs. 2. Acts directly on arterial/vascular SM to increase total peripheral resistance 3. Stimulates secretion of aldosterone |
|
|
Renal response to low blood pressure/volume
|
- low pressure and/or afferent arteriole constriction is sensed as ↓glomerular pressure → ↓GFR → ↓Filtered load → ↑Prox tube efficiency (more time/area) → ↑Na reabsorption
- In the distal nephron: ↑ADH (pituitary) + ↓Na (stronger gradient) → ↑Na reabsorption + ↑H2O reabsorption → (secrete renin) → ↓urine volume |
|
|
Baroreceptors comminucating with the kidney
|
Baroreceptors = physiological pressure gauges
- High pressure: in carotid sinuses and aortic arch; sense arterial pressure. - Low pressure: in cardiac atria and pulmonary vessels; sense fullness of vasculature. - Intrarenal: in afferent arterioles; sense renal artery pressure. |
|
|
Aldosterone Action and regulation
|
= mineralocorticoid produced by adrenal glands (via renin/angiotensin stimulation). Acts on principal cells of CD's to increase Na reabsorption (H2O retention) (also excrete K)
Upregulated by: ↑[Angiotensin II], ↑[K], ↓[Na] (↓vol/P mostly by AngII), ↑ACTH (stims adrena medulla) Mechanism of action: receptor inside cells, binding Aldo it travels to nucleus as a Tx factor, increases proteins for Na reabs (ENaC channel). [Other steroid hormones can bind, 11B-HSD is competitive receptor to prevent wrong activation) Action corrects: ↓Na/BP: ↑H2O reabsorption, ↑BP ↑[K]: ↑K transport from ECF via Na/K exchanger, ↑K excretion by luminal channels |
|
|
Bicarbonate reabsorption in the kidney
|
- kidney must reabsorb nearly all bicarbonate (diet usually leaves acid surplus)
Prox tubule: 75% abs; Na/H+ exchanger channel put H+ in lumen where it binds HCO3, CO2 diffuses into cell, degraded via carb.anhyd., HCO3 moved to interstitium by Na symporter. No effect on pH - In CD: type A intercalated cells excrete H/reabsorb base (type B to opposite). CO2 inside cell is split via C.A., H+ pumped to lumen via H/K channel + H-ATPase; HCO3 reabsorbed by Cl/HCO3 transporter. This increases urine pH --Mechanism flipped to excrete HCO3 - HCO3 can be "generated" whenever a different buffer is available to accept H+ in the lumen |
|
|
Renal response to acid/alkali load
|
Acid load: surplus of H+ so kidney will work to secrete it (utilize type A cells in the CD)
Alkali load: surplus base, so will excrete it (bicarbonate); utilize type B cells in CD. (CO2 diffuses in, split, HCO3 to lumen via Cl swapper, H+ to interstitium by K swap) |
|
|
Phosphate as an alternate renal buffer
|
- At normal pH, 80% exists in the conjugate base form (HPO4)
- Total filtered load is about 160mmol/day. Most (75-90%) is reabsorbed in the PT and about 40mmol is available for buffering. - Each H+ that combines with a P04 releases a molecule of HCO3 into the bloodstream. |
|
|
Ammonium in renal buffering
|
- formed in the liver from protein catabolism. At high pH converted to urea and excreted, at low pH bound w/ HCO3 as glutamine
- glutamine converted back in PT--net gain HCO3, NH4 travels to the lumen and is excreted - 59mmol/day available to buffer (in acidic conditions) - In the medulla, NH4 travels to interstitium via NKCC, converted to NH3, reabsorbed by Rhcg/Rhbg transporters in CD to accept H from HCO3 |
|
|
Animal models of glomerulonephritis
|
Acute experimental serum sickness:
- Antibodies: preformed circulating, single antigen - LM: diffuse proliferative GN (>50%), monocyte infiltrate, endothelial swelling - EM: mesangial, subendothelial, subepithelial (“humps”) dense deposits, no spike/dome formation since short process - IF: starry sky IgG pattern from mesangial deposits and subepithelial humps (punctate areas) - Human: acute post-streptococcal GN Heymann NephritiS: - preformed against megalin in foot processes - LM: FP effacement - EM: subepithelial dense deposits with relative periodicity, spike/dome formation (chronic) - IF: granular IgG - Human: idiopathic membranous nephropathy (human target is phospholipase A2 receptor) Masugi Nephritis: - preformed antibodies against heparan sulfate - LM: crescentic GN - EM: no dense deposits visible (not condensed) - IF: linear IgG - human: anti-GBM, Goodpasture’s disease (target NC1 domain of collagen IV) |
|
|
Complement in glomerular injury
|
Classical pathway:
- activated in type III hypersensitivity response (SLE, some GN, acute serum sickness): immune complex deposition results in C3 activation and cleavage in to C3b which is the central component of the membrane attack complex formatio→ direct cell/tissue injury via formation of transmembrane channels (lytic pores), leukocytes also recruited Alternative pathway: - antigen deposition in the membrane leads to complement activation and phagocytosis, also leukocyte recruitment-→glomeruloneprhitis --Complement is the main driver of GN |
|
|
Factors that influence immune complex deposition
|
Antigen immunogenicity: not all antigens elicit and antibody response
Antibody class: usually IgG, occasionally IgM/A Immune complex size: large circulating complexes are efficiently cleared by monocyte phagocytic system (MPS), while intermediate ones (~500kDa) are cleared less and are more likely to deposit in glomeruli Immune complex avidity and charge: determines whether can dissociate to pass through GBM or are taken up by mesangial cells - subepithelial: low avidity + positive charge (attracted to and pass through filtration barrier) - mesangial: high avidity + neutral charge - subendothelial: low avidity + neutral charge - not deposited: high negative charge (since repelled by GBM) |
|
|
Histologic exams for diagnosing immunologic causes of glomerular injury
|
- electron, light, and fluorescence microscopy
- FM: can differentiate with staining between different immunoglobulins (IgG/M/A) and screen for complement involvement (C3, C1q) - EM: can visualized dense deposit location within the glomerulus (relevant to antigen-antibody affinity) and predict chronicity (based on glomerular response) - LM can show changes in different parts of the glomerulus—hetero/homogeneous changes |
|
|
Mechanisms mediating immunologic glomerular disease
|
In these diseases antibody binds to antigen forming a complex either fixed in the GBM or circulating that lodges in the GBM/mesangial matrix initiating a hypersensitivity response by the glomerulus
Type II (antibody-mediated) hypersensitivity: - involves production of IgG/M which binds to antigen on target tissue/cell leading to phagocytosis or lysis through complement activation or leukocyte recruitment. - typically in-situ immune complex formation, can lead to diffuse deposition not visible by EM (since antigen is fixed/can’t aggregate) - Eg: Goodpasture syndrome (attacks collagen domain in GBM) Type III (immune complex mediated) hypersensitivity: - involves deposition of antigen-antibody complexes in the GBM leading to complement activation, leukocyte involvement, and release of enzymes and inflammatory mediators. - Typically circulating/plasma soluble, preformed complexes deposit in glomerulus, location based on complex affinity - Eg: SLE, some types of GN, serum sickness, arthus reaction |
|
|
Mechanisms of glomerular injury
|
- Immunologic diseases (resulting in glomerulonephritis or glomerulopathy)
- Metabolic diseases (Diabetes mellitus, amyloidosis) - Coagulation disorders (DIC, HUS, TTP) - Hypertension - Congenital/hereditary diseases - Infectious - Idiopathic |
|
|
Congenital Nephrotic Syndrome (Finnish-Type)
|
= onset of nephrotic syndrome (massive proteinuria w/ edema) within the first 3 months of life, usually within a few days
- Associated w/ premature birth, large placenta, skeletal deformities, poor motor development, progressive renal failure - seen most commonly in Finland - Autosomal recessive, linked to mutations in NPHS1 gene (encodes nephrin in the nephrin zipper) - resistant to most treatments (bilateral nephrectomies sometimes indicated in severe cases) - death secondary to sepsis or other complications (hypercoagulopathy, infection, stroke—mostly ) |
|
|
Molecules conferring anionic basement membrane charge
|
Conferred by two molecules:
- Podocalyxin: coats the surface of podocytes foot processes - Heparan sulfate: occurs as tiny aggregates (“anionic sites”) in the basement membrane (lamina rara interna/externa) |
|
|
Nephrotic syndrome definition
|
1. severe proteinuria (>3.5g/day)
2. Reduced serum albumin 3. Edema 4. Hyperlipidemia 5. Hyperlipiduria (see oval fat bodies on LM) - damage to kidneys has opened filtration barrier to allow passage of protein (but not RBCs) - creatinine will be normal because still cleared effectively Ex: membranous GN, minimal change disease, amyloidosis, diabetic GN, focal segmental GN, membranoproliferative GN |
|
|
Structure of the glomerular basement membrane
|
- consists of meshwork of collagen IV and proteoglycan fibrillae with spaces for water and small solutes to filter
3 layers: - lamina rara interna (LRI) electron lucent, filters by charge (heparan sulfate) - lamina densa (LD) electron dense, filters by size (MW < 5800kDa) - lamina rara externa (LRE) electron lucent, filters by charge (heparan sulfate) foot processes partially embedded |
|
|
Glomerulus cell types
|
- Capillary endothelial cells: fenestrated (excludes large proteins and cells), provide first filtration
- Podocytes: sit on outside of GBM, foot processes interdigiate to form slit pores (connected by nephrin zipper—proteins linked to actin in FPs) creating apertures of ~40A (but can contract w/ actin to change diameter). Direct communication allows for signals to be rapidly transmitted across the entire glomerulus. - Mesangial cells: form arborizing stalk by secreting mesangial matrix (forms scaffolding for proteins); moderates blood flow/filtration in the glomerulus via contraction of actin/myosin; can migrate to capillary loop to remove immune complexes from the GBM; secrete prostaglandins and other immune modulatory molecules. Proliferate with damaged Parietal epithelial cells: line inside of bowman’s capsule JgA cells: regulate RAAS via secretion of renin. Work in conjunction with macula densa cells |
|
|
Calcium alkali syndrome
|
- results from ingestion of large amount of calcium and absorbable alkali with resulting hypercalcemia: overdose of calcium citrate or calcium carbonate (tums).
- Pathogenesis: excess base causes anion drag increasing Na excretion, and causing volume loss , Cl sensitive alkalosis, and calcium retention. PTH is also suppressed increasing base retention. Ca Retention + excess intake causes Ca to precipitate, particular in the kidney resulting in acute kidney injury (worsening volume depletion and alkalosis. |
|
|
Chloride-resistant metabolic alkalosis
|
Mineralcorticoid excess:
- primary hyperaldosteronism, Cushing’s, ectopic ACTH, secondary hyperaldosteronism (renovascular disease, malignant HTN, CHF w/ diuretics, cirrhosis w/ diuretics) - aldosterone stimulates Na/K exchange in the distal nephron, resulting in K wasting. Low serum [K] stimulates ammonia production (from glutamine) resulting increase H+ excretion. Aldosterone also stimulates acid secretion via H+ATPases - urinary Cl normal due to volume expansion/HTN→ ↑Na/Cl filtration which suppresses JGA and blocks Cl reabsorption - Will not respond to saline, must correct underlying cause Hypokalemia: any cause - as above, will stimulate NH3 production from glutamate, increasing renal acid excretion |
|
|
Renal tubule acidosis Type II
|
= mild renal falure causing acidosis due to impaired bicarb reabsorption in the Proximal tubule/impaired NH4/H2PO4 excretion.
Characteristics: requires high does HCO3 to treat (10-15 mEq/kg/d), variable urine pH (distal H+ secretion still working so usually less severe acidosis), serum HCO3 usually 14-20, normal/low plasma K. Often associated with Fanconi syndrome (generalized proximal tubule dysfunction) Causes: cystinosis,tyrosinemia, galactosemia, glucose storage disease type I, carbonic anhydrase inhibitors (acetazolamide), others |
|
|
Renal tubule acidosis Type IV
|
= mild non-tubular renal acidosis, due to physiologic reduction in ammonium excretion secondary to hypoaldosteronism
Characteristics: hyperkalemia, urine pH <5.3, serum HCO3 >15, corrected with 1-2 mEq/kg/d HCO3 (or none if K+ okay) Mechanism: hypoaldosteronism leads to Na wasting/K retention, high serum [K] results in reduced ammonia production from glutamine (normally high K in ammonia containing foods—body thinks large amounts of ammonia consumed) and lessens distal H secretion Causes: medication (anti-hypertensives) |
|
|
Renal net acid excretion
|
NAE = NH4 + TA
Titratable acidity (TA) = H + H2PO4 – HCO3 NAE usually 10,000–150,000nM/L, with 300–10,000nM/L H+, for a final pH of 5-6.5 |
|
|
Chloride sensitive metabolic alkalosis
|
= alkalosis with decreased arterial blood volume, and urine Cl <20mEq/L, urine Na also usually low (except w/ diuretics)
Pathophys: Low arterial blood volume results in RAAS activation (sympathetic/osmotic stimulation) and Na retention (↑Na/H exchanger activity). Cl is passively reabsorbed w/ Na, resulting in ↓[Cl] in the distal tubule which therefore reduces Cl/HCO3 exchanger function, reducing HCO3 excretion. Aldosterone also stimulates acid secretion via collecting duct H+ATPases. Low Cl maintains JGA stimulation for renin release maintaining alkalosis Causes: Bicarbonate load with bicarb/anion drag (acute alkali administration, Milk-alkali syndrome, recovery from AG acidosis), Effective volume contraction (GI losses—vomiting, NG suction, congenital chloridorrhea, villous adenoma; Renal losses: diuretics, post-hypercapnic state, hypercalcemia, non-reabsorbable anoin (cabenicillin, sulfate), magnesium deficiency, Bartter’s syndrome) Treatment: saline, correct cause of hypotension |
|
|
Renal tubule acidosis Type I
|
- indirectly measures ammonia present in urine (can’t be detected clinically). Used to distinguish renal/non-renal causes of acidosis
- In acidosis extra NH3 (produced from glutamine in the proximal tubule) should be binding H+ in the distal tubule to allow for HCO3 reabsorption. - In non-renal acid a urine will have high [NH4+] and [Cl-] to balance, while [Na] and [K] are constant resulting in large negative anion gap (<-20) - In renal acidosis, impaired renal function means reduced ammonia production and H+ binding, so low urine [NH4+] and [Cl-], resulting in an anion gap that is zero or slightly positive - Not useful parameter of high AG acidosis with renal loss of anions (such as diabetic ketoacidosis). Also not useful in volume depleted states due to Na retention |
|
|
Urinary anion gap
|
= failure of the distal tubule to secrete H+ (failure to acidify urine)
Characteristics: urine pH >5.3 (despite acidemia), serum HCO3 may by <10, plasma K usually low, treatment achieved with 1-2 mEq/kg/d HCO3, possible calcium phosphate stones and nephrocalcinosis, can results in hypercalciuria and bone demineralization (rickets, osteomalacia) Causes: usually due to interstitial nephritis caused by lead, lithium, analgesic abuse, autoimmune conditions (Sjogren’s, SLE, PBC), recessive mutations in Cl/HCO3 exchanger, drugs (amphotericin B—antifungal creates wholes in basement membrane allowing H+ back leak) Etiologies: multiple ex: apical H+/K+ exchanger dysfunction, apical H ATPase pump dysfunction, leaky membrane causing back flow of H+ |
|
|
Metabolic Acidosis Pneumonics
|
High Anion gap: MUD PILES
Methanol Uremia due to chronic renal failure Diabeteic ketoacidosis Propylene glycol Infection, iron, inborn errors of metabolism Lactic acidosis Ethylene glycol (anti-freeze)/Ethanol Salicylates Normal anion gap: HARD UP Hyperalimentation Acetazolamine (carbonic anhydrase inhibitor) Renal tubular acidosis Diarrhea Uteroenteric fistula Pancreaticoduodenal fistula |
|
|
4 Steps to understanding acid-base disorders
|
1. Obtain ABG and serum electrolytes
2. Determine internal consistency (if data is any good): use CO2 and HCO3 to calculate [H], see if matches measured value. [H+] = 24 x (CO2/HCO3) (use plasma CO2). Defects may be due to air exposure, processing time, poor storage 3. Guess primary disorder (metabolic/respiratory acid/alkalosis, simple vs. mixed disorder): look at pH (acid/alk), the pCO2 or bicarb (resp/met) 4. Calculate anion gap and determine if compensation is appropriate: AG = Na+ – (HCO3- + Cl-) normal around 10mM. The use compensation formulas |
|
|
Models for acid/base disorders
|
1. Bicarbonate model: pH, HCO3, pCO2 dissolved in the blood as the main buffer system, look at anion gap for imbalances
2. Base excess model: use base excess instead of [HCO3]. Often used in the OR and ICU for anesthesia 3. Strong ion difference: variables derived from pH, pCO2, electrolytes, and lactate. Complicated (requires a computer to calculate) |
|
|
Consequences of pH imbalance
|
Acute mild acidosis (to 7.1): tolerated well, can be adaptive to increase O2 delivery by Bohr effect and vasodilation (good for exercise)
Acute severe acidosis (<7.1): problems with protein folding, ventricular arrhythmias, catecholamine resistance (maladaptive for shock as it lowers BP), <6.8: lethargy, coma, ARDS Chronic acidosis: may arise from standard western diet (protein deficient—used for ammonia) leading to catabolism of musclem (muscle atrophy), increase susceptibility to infection, negative calcium balance and calciuria (osteoporosis, nephrolithiasis) Acute/chronic alkalemia: reduced ionized calcium leading to tetany and cardiac arrest (>7.75), decrease tissue oxygenation + compensatory hypoventilation (causes mortality in the acutely ill) |
|
|
Acid/base balance
|
Intake:
- carbonic acid (CO2 from metabolism) and non-carbonic acid(sulphates, phosphates from diet) - organic bases: citrate, acetate, acetic acid (from fruits and vegetables) converted to base in liver Excretion: - fecal: bicarb (makes blood more acidic) - respiration: excretes carbonic acid (CO2) - renal: bicarb secreted or reabsorbed, H+ secreted (Na/H antiporter) allowing bicarb reabsorption, ammonia also accepts protons for excretion |
|
|
Immunosuppression regimen in kidney transplant
|
Induction:
- anti-thymocte globulin - daclizumab (anti-IL2) Maintenance: - Tacrolimus (calcineurin) - Mycophenolate mofetil (cytoxin) - Prednione (glucocorticoid) Cellular rejection: - mild: prednisone (3 days) - severe: anti-thymocyte globulin (7-10 days) |
|
|
4 Main mechanisms of immunosuppressants
|
- T-cell depletion: anti-thymocyte globulin, muronomab-CD3
- T-cell activation: Cyclosporin, Tacrolimus, Abatacept/Belatacept - Cytokines (synthesis, signaling): Sirolimus, daclizumab, glucocorticoids - Proliferation: mycophenolate mofetil, glucorticoids |
|
|
Prednisone (immunosuppressant)
|
Class: glucocorticoid
- affects lymphocyte trafficking (anti-adhesion preventing lymphocyte extravasation from circulation - inhibits T-cell proliferation - inhibits cytokine expression/signaling - inhibits macrophage and lymphocyte function Mechanism: - binds to intercellular steroid receptors so can effect transcription of large gene groups. Stimulating the production of IκB which binds and sequesters NF-κB in the cytoplasm so it cannot travel to the nucleus to activate transcription of genes during immune response (incl: enzymes, adhesion molecules, cytokines). Effects multiple points in activation pathway |
|
|
Mycophenolate mofetil
|
Class: cytotoxin
Mechanism: inhibits inosine monophosphate dehydrogenase (IMPDH) preventing the conversion of IMP to XMP and reducing de novo systhesis of GTP (lymphocytes rely on this pathway more than other cell types when proliferating). Reduced GTP reduces DNA synthesis preventing production of cytotoxic T cells from activated CD8 cells - Given as pro-drug, produces active metabolite—mycophenolic acid |
|
|
Daclizumab (Zenapex)
|
Class: antibody reagent
= mouse monoclonal antibody against CD3 on T-helper cells Mechanism: binds to CD3 on T-helper cells preventing activation by APCs. Results in decrease in T-cells |
|
|
Muromonab-CD3 (OKT3)
|
Class: antibody reagent
= genetically engineered Anti-IL2 receptor antibody Mechanism: binds to IL-2 receptor on CD8 cells preventing cytokine-mediated activation (some minor B cell effect) |
|
|
Anti-thymocyte globulin
|
Class: antibody reagent
= purified IgG produced by horses in response to injection of human lymphocyte Indication: used before transplant to reduce the total number of lymphocytes available to mount initial immune response to transplanted organ Mechanism: induces complement deposition on T-cells resulting in cell death Side effect: excess response to drug can lead to complement depletion |
|
|
Abatacept/Belatacept
|
Class: T-cell suppressants/costimulatory blockade
Abatacept for RA, Belatacept for transplant (2AA substitution makes it 10x more potent) Mechanism: modified CD28 receptors (on antibody heavy chain making it soluble) bind B7 on APCs but not MHC so cell goes into anergy from single signal. Net effect is to block activation of the T-helper cell by the APCs, preventing activation of both CD4 and CD8 T-cells Side effects: anemia, diarrhea, UTI, peripheral edema, constipation, HTN, pyrexia, graft dysfunction, cough, nausea/vomiting, potassium dysfunction, leukopenia |
|
|
Sirolimus (rapamycin)
|
Class: T-cell suppressant
Mechanism: binds to FKBP12 (same receptor as tacrolimus) then binds and inhibits mTOR (mammalian target of rapamycin) to prevent signal proliferation via p70s6 after cytokine binding (normally would result in DNA synthesis and cell proliferation). Net result is to inhibit activation of CD8 T cells or B cells by prevention of cytokine signaling/activation Side effects: difficulty wound healing—mouth ulcers, drug interactions (p450s), long half-life (72hrs) so less nephrotoxic than calcinuerin inhibitors |
|
|
Calcineurin Inhibitors
|
Class: T-cell suppressants, eg: Cyclosporin A (binds cyclophilin), Tacrolimus (binds FKBP12)
Mechanism: receptors complex inhibits calcineurin (normally activated by Ca++ influx via PLCγ after antigen binding) preventing dephoshorylation/activation of NFAT (nuclear factor of activating T cells). NFAT therefore cannot enter the nucleus to pair w/ AP1 (via PLCγ→PKC→MAP kinase) and induce transcription of cytokines (IL-2, IL-4, GM-CSF). Overall prevents activation of helper T-cells, preventing activation of CD8 and CD4 T cells Side effects: - main: hypertension, renal dysfunction, diabetes, gingival hyperplasia. (now small molecules being develop to selectively target NFAT binding site on calcineurin to reduce effects) - others: hyperkalemia, hyperlipidemia, tremor, hyperglycemia, nephrotoxicity, hirsutism, alopecia (tacrolimus), drug interactions (since metabolized by Cyt p450) |
|
|
4 Classes of immune suppressants
|
- T-cell suppressants: cyclosporine A, Tacrolimus (FK506), Sirolimus (rapamycin), Abatacept/Belatacept
- Antibody reagents: anti-thymocyte globulin, Muromonab-CD3, Daclizumab - Cytotoxic drugs: Mycophenolate mofetil - Glucocorticoids (prednisone) |
|
|
Side effects of long term immunosuppression
|
- infection: immediate risk for susceptibility to foreign organisms
- malignancy: long term risk. Immune surveillance theory: abnormal cells arise spontaneously, the immune system normally eradicates them before they can proliferate into cancer |
|
|
Indications for immunosuppression
|
Organ transplant
Autoimmune disease Cancer (prevent proliferation of cells) |
|
|
Renal transplant criteria
|
Recipient
Pre-transplant evaluation: med Hx, PE, CBC & chemistries, PTT, PPT, blood typing, Hep B/C, HIV, CMV, pelvic and PAP, CXR, EKG, HLA tissue typing, venereal disease screen - R/o contraindications: malignancy, cirrhosis, severe myocardial dysfunction, active mental illness, chronic infection, acute substance abuse, extreme obesity (BMI >35) Post-transplant therapy for: active infection (Hepatitis, TB, CMV), CV disease, PUD, cerebrovascular disease, substance abuse Live Donor: adult (>18), no contraindications: HTN/pre-HTN, diabetes, proteinuria, GFR<80mL/min, microscopic hematuria, multiple renal vessels/anomalies, significant medical illness, history of thrombosis/thromboembolism, strong fam Hx of renal disease diabetes or HTN, pregnancy |
|
|
Ureteral ectopia
|
= the ureter terminates at a different site than the urinary bladder
- associated with renal dysplasia, UTIs, incontinence Mechanism: inability of the elongating ureteric bud to incorporate into the bladder wall (most commonly remains fused with the urethra Incidence: - men: 47% prostatic urethra, 33% seminal vesicle, 10% prostatic utricle, 5% ductus deferens, 5% ejaculatory duct - women: 36% urethra, 34% vaginal vestibule, 25% vagina, 5% urterus or cervix |
|
|
Horseshoe kidney
|
= fusion of the kidneys in a horseshoe shape
Incidence: 1:400-600, sporadic Mechanism: fusion of the lower (95%) or upper (5%) poles of the kidney during development. Obstructs rotation so the renal pelvis faces anteriorly and migration is blocked by the inferior mesenteric artery Sx: normally asx, discovered incidentally Complications: slight risk of infections and renal pelvis tumors, Wilm’s tumor |
|
|
Renal ectopia
|
= atypical position of the kidney
Incidence: 1:900, sporadic Etiologies: simple (pelvic, subdiaphragmatic, thoracic) or crossed (+/- fusion). Associated with other GU malformations. Increased risk of UTI (due to stasis) and renal injury (not protected) Mechanism: abnormal migration of the kidneys from the pelvic region (L4) to the retroperitoneum (T12/L1) |
|
|
Supernumerary kidneys
|
= development of >2 kidneys
Mechanism: splitting of the nephrogenic blastema leading to formation of partial or completely duplicated ureteral stalks that become separate enacapsulated kidneys Also, duplication of ureters = multiple ureters emptying from a kidney Mechanism: duplication or branching of a uteric bud |
|
|
Renal hypoplasia
|
Renal hypoplasia = kidneys with <6 pyramids (normal >10), kidney weight <50% expected
Incidence: very rare (renal atrophy much more common) Sx: unilateral (asx), bilateral (variable degree of renal insufficiency and HTN) Etiology: renal artery atherosclerosis |
|
|
Potter’s syndrome
|
= secondary anaomaly of bilateral renal angenesis
Mechanism: absemt fetal urine → reduced amniotic fluid excretion (oligohydramnios) Sx: - characteristic facial features: beak nose, skin folds under eyes, flat and low-set ears - limb deformities - pulmonary hypoplasia - amnion nodosum (nodules on the fetal surface of the amnio) - results in death in utero or shortly after birth from renal or pulmonary insufficienct |
|
|
Renal agenesis
|
Incidence: unilateral (1:1000, M>F), bilateral (1:4000, M>F), sporadic
Mechanism: ureteric bud does not form or fails to induce differentiation of the metanephric blastema Sx: - unilateral: normal function, but glomerulosclerosis may be a late complication - bilateral: Potter syndrome. Death in utero or shortly after birth form renal or pulmonary insufficiency |
|
|
GDNF-RET signaling system
|
GDNF = glial cell-derived neurotrophic factor, C-Ret (RET) = a receptor tyrosine kinase (RTK)
- primary purpose of GDNF-RET in kidney development is to induce ureteric bud growth from the mesonephric duct (GDNF is produced by blastema cells) Steps in pathway: (KO of any signaling molecules can cause serious dysplastic GU development or kidney agenesis (RET KO)) 1. GDNF binds to GFRα1 forming a complex that acts as a ligand and binds the RET receptor 2. Binding activate RET dimerization and cross-phosphorylation of Tyr residues and SH2 domains 3. Activated SH2 domains recognize and bind specific motifs on downstream intracellular proteins (eg. Tyr1096) leading to transcriptional activation 4. Activated GDNF-RET complex can activate more complexes creating signal cascaded to activate transcription factors |
|
|
Embryogenesis of the bladder and uretha
|
- endodermal cloaca is divided by the urorectal septum to form the urogenital sinus and the rectum.
- the baldder develops from the vesicular part of the urogenital sinus - in in females the pelvic part of the urogenital sinus becomes the entire urethra. In males the pelvic part becomes the prostatic while the spongy (penile) part develops in the phallus. Most distal part of the male urethra is ectodermal - The expanding bladder wall incorporates the ureteric bud (growing off the mesoneprhic duct), flaring out to form the trigone of the bladder |
|
|
Embryogenesis of the ureter, renal collecting duct and nephrons
|
- Intermediate mesoderm (between paraxial and lateral plate) forms the pronephros (rostral), mesonephros, and metanephros. Pro and mesonephros form rudimentary kidneys then mostly degenerate.
- mesonephrotic duct (through mesonephros to cloaca) forms the Wolfian duct which forms the male genital ducts (epididymis and ductus deferens) and regresses in females) - Metanephros begins a diverticulum off mesonephrotic duct w/ metanephric mass (containing metanephric blastema cells→ nephrons) and develops into the adult ureter, renal pelvis, calices, and collecting tubules |
|
|
Foods high in K
|
Recommended intake: 120mmol/d (normal), or 51mmol/d (end-stage renal failure)
- avocado, artichoke, tomato paste, beef, potato, squash, raisins, prunes, cantelope, orange juice, banana |
|
|
Treatment of potassium disorders
|
Hyperkalemia:
- IV calcium gluconate (over CaCl-central line, tissue necrosis) for cardiac membrane stabilization. Do not mix w/ HCO3, care w/ digoxin (lead to toxicity) - insulin or β-2 agonists to treat redistribution. Care in patients w/ risk factors for tachycardia, ischemia - kayexalate, hemodialysis for direct K removal Hypokalemia: - oral KCl for diuretic or volume depletion. Micro-encapsulated has lowest complications. If IV, use 10mmol/hr, if faster use central line + continuous EKG monitoring. Typically 20mmol KCl→ ↑0.25mmol/L [K] - K-phos if also phosphorus depleted - KHCO3 for acidosis - KCitrate for renal stone prevention - If refractory, may have Gitelman, obstain plasma magnesium |
|
|
EKG changes associated with potassium disorders
|
Hyperkalemia:
- peaked T waves (starts around 6-7), flattened P waves (7-8), prolonged PR interval (7-8), atrial standstill (8-9), depressed ST segments (8-9), widened QRS complex, sine-wave patterns (>9, severe) Hypokalemia: - U waves (3-3.5) +ST depression (2.5-3) + decreased T wave and irregularity/inversion (3-3.5) = “rollercoaster effect,” (<2.5) decreased/widened QRS, normal QT, increased P, prolonged PR |
|
|
Hypokalemia workup
|
1. obtain urine, if [K]< 20mmol/d non-renal loss (vomiting, diuretic use, poor intake) which should be corrected
2. if [K] >20mmol/d (renal loss), calculate TTKG. If TTKG<2 → increased tubular flow or osmotic diuresis. Correct 3. If TTKG >4 determine acid/base status. If acidosis: distal (type I) RTA, proximal (type II) RTA, ketoacidosis, amphotericin B, acetazolamide 4. If alkalosis check BP: - high: mineralcorticoid excess, Liddle’s syndrome - low or normal: loop or thiazide diuretic, vomiting, gastric suction, Barter syndrome, Gitelman syndrome |
|
|
Work up for hyperkalemia
|
1. re-run labs (r/o pseudohyperkalemia, eval for hemolysis and elevated leukocytes and platelets), evaluate EKG (check for abnormalities—if yes, emergent therapy required)
2. Locate source of hyperkalemia: transcellular shift (trauma/cell lysis/necrosis), K supplement, medications (ACE, ARB, NSAIDs, K-sparing diruetics), acute kidney injury (oligouria, GFR<20mL/min/1.73m2) 3. If none of the above evaluate transtubular K gradient: TTKG = (Ku/Kp)x(Posm/Uosm); normal 6-12. - >7 non-renal cause: decreased effective circulating volume - <5: dose w/ 0.5mg 9α-Fludrocortisone. No change→ medication induced receptor blockade or tubular mineralcorticoid resistance. If responds (TTKG >10) then 1°/2° hypoaldosteronis |
|
|
Manifestations/severity of hyper/hypokalemia
|
Manifestations vary depending on severity and speed of onset. Most are because it alters the inter/extra cellular gradients preventing normal function (esp in muscle cells)
- Classic: EKG abnormalities (always concerning), weakness, if severe respiratory failure from diaphragm paralysis Severity: - <2.0: ascending paralysis, respiratory impairment - <2.5: muscle necrosis risk - 2.5-3: weakness constipation - 3.0-3.5: often asymptomatic unless at risk for cardiac arrhythmias - 3.5-5.0: normal - 5.1-5.5: borderline, often tolerated well unless acute rise - 5.6-5.9: often tolerated will if chronic, need to work up - 6.0-6.9: requires prompt evaluation and frequent monitoring - >7.0: Life threatening, ICU monitoring |
|
|
Hypokalemia causes
|
= plasma [K] > 3.5mmol/L
- decreased intake: starvation - redistribution: insulin excess, β-adrenergic catecholamines, pseudohypokalemia (large # abnormal leukocytes) - non-renal K loss: diarrhea, vomiting, severe burns, profuse sweating - renal K loss: diuretics, mineralcorticoid excess, renal dysfunction - hormonal: hyperaldosteronism, Liddle syndrome, Gittleman’s syndrome |
|
|
Hyperkalemia causes
|
= plasma [K] > 5.0mEq/L (differentiate from pseudohyperkalemia—artifact from K release from cells prior to analysis)
Causes: - redistribution: insulin deficiency, solvent drag, non-organic/mineral metabolic acidosis(↑[H] → + charge on cells→ K displaced to charge balance) , respiratory acidosis, iatrogenic/meds (statins, cocaine, chemo, succinylcholine), ongoing release from cells (rhabdomyolysis, tissue necrosis, tumor lysis, large hematoma) - reduced excretion: decreased effective circulating volume (less filtered/secreted), 1°/2° hypoaldosteronism (impaired K secretion following defective ENaC reabsorption—reduced Na/K ATPase activity to charge balance), tubular mineralcorticoid resistance, drugs (digoxin, calcineurin inhibitors-cyclo, tac) - increased intake: usually only if kidney is also abnormal—normally handle up to 400mmol/day w/ <1mmol/L K increase in plasma (normal intake >100mmol/d) - RAAS derangement: reduced Na intake results in reduced need for Na/K ATPase to balance charge, so less is excreted; drugs (β-blockers, NSAIDS/COX-2, ACE-I, ARBs, heparin, spironolactone, eplerenone, amiloride, trimethoprim, pentamidine) |
|
|
Na vs H2O retention
|
H2O: dependent on ADH release/regulation from the posterior pituitary, which causes reabsorption in the distal nephron independent of Na (aquaporins). Secondary (non-renal) retention from low serum oncotic pressure (causing interstitial accumulation)
- stimuli: osmotic (high serum osmolality), - pathologic states: hyponatremia, edema (venous obstruction, decreased oncotic pressure) Na: dependent on aldosterone release/regulation from the adrenal glands (ENaC) - stimuli: low volume (CHF, cirrhosis) resulting in RBF/GFR stimulating the JG cells to release renin (b/c low filtrate [Na]) - pathologic states: hypernatremia, edema (b/c Na retention draws H2O), hypertension |
|
|
Edema pathophys
|
= manifestation of excess Na accumulation in the body
2 causes: (ultimately resulting in fluid extravasation in excess of lymphatic drainage, causing accumulation) - Na/H2O retention by the kidney in response by perceived/effective reduction in ECFV (reduced renal perfusion due to ↓CO, splanchnic vasodilation, hypoalbuminemia) via activation of RAAS (results in increased circulating volume and hydrostatic pressure) and production of local vasoconstrictors (NE, AngII—reduce GFR. Usually initial cause of - Alteration of capillary hemodynamics to reverse starling forces (oncotic, hydrostatic pressures: hypoalbuminemia, increased circulating volume/Na) favor movement of solute into the interstitial space. Usually later state following renal retention Net filtration (across capillary barrier): LpS(Pcap – Pif) – σ(Ocap – Oif) where LpS is barrier permiablity/filtration coefficient and σ is the reflection coefficient |
|
|
Diagnosing Acid-base disorders
|
Identify pH abnormalities: (determine if metabolic/respiratory, acid/alkalosis)
- <7.35, acidosis: if ↓[HCO3]→metabolic; if ↑Pco2 →respiratory - >7.45, alkalosis: if ↑[HCO3]→metabolic; if ↓Pco2 →respiratory - If normal: look for abnormal Pco2 or HCO3 and proceed from there (normal Pco2 = 40mmHg, HCO2 = 24-26mEq/L) Determine if compensation is correct: - metabolic acidosis: Δ pCO2 = 1.2[Δ HCO3] - metabolic alkalosis: Δ pCO2 = 0.7[ΔHCO3]) - Respiratory acidosis: acute— ↑ Δ[HCO3]= 0.1(ΔpCO2); chronic— Δ[HCO3]= .35(Δ pCO2) - Respiratory alkalosis: acute— ↓ Δ[HCO3]= 0.2(ΔpCO2); chronic— Δ[HCO3]= 0.5(ΔpCO2) Calculate anion gap: AG = [Na+] – ([Cl-] + [HCO3-]) - normal 9-16mEq/L; AG>20 likely acidosis, AG>30 certainly high AG acidosis Compare Δ ↑AG/ Δ↓[HCO3]: if 1.5→lactic acidosis; if 1→ketoacidosis - if measured [HCO3] is ↑ for AG→hidden M. Alk; if [HCO3]↓ for AG → hidden M. Acid |
|
|
Treatment for metabolic alkalosis
|
Identify and treat underlying inciting factor and cause for renal retention
ECFV depletion: replacement with NaCl .9% saline (for chronic diarrhea proceed carefully) K depletion, Mineralocorticoid excess, Bartter’s/Gittelman: KCl replacement Alkalosis associated with ECFV overload/renal failure: NaCl contraindicated, K infusion potentially dangerous; dialysis may help |
|
|
Respiratory Compensation for metabolic alkalosis
|
[H+] α Pco2/[HCO3]
- as ↑[HCO3], respiration slows to ↑Pco2 and maintain [H+] - After compensation Pco2 = 40 + 0.7 x ([measured HCO3] – [normal HCO3]); normal HCO3= 24-26mEq/L - >5mmHg deviation in Pco2 from expected compensated value means concurrent respiratory disorder: ↑Pco2→ respiratory acidosis, ↓Pco2→respiratory alkalosis |
|
|
Metabolic alkalosis
|
- increase in plasma pH generated by either loss of H+ or increase in HCO3, maintained by abnormal renal retention of HCO3
Etiologies: - ECFV depletion (chloride depletion syndrome (low urine Cl), saline-responsive): vomiting/NG-suction (loss gastric acid), diuretic therapy, post-hypercapnea, chronic diarrhea/laxative abuse - severe K depletion (saline-resistant): any cause, must be replaced before treatment - Mineralocorticoid excess syndromes (saline-resistant): primary hyperaldosteronism, Cushing’s, ectopic ACTH, secondary hyperaldosteronism (renovascular disease, malignant HTN, CHF w/ diuretics, cirrhosis w/ diuretics) - Gitelman’s syndrome (saline resistant): loss of thiazide sensitive Na/Cl symporter - Bartter’s syndrome (saline resistant): loss of NaKCl2 transporter - Secondary to post-chronic hypercapneic state: kidney compensates for acidosis but cannot return to normal once acidosis is corrected due to renal failure - Metabolic alkalosis maintained by renal failure (saline generally contraindicated) |
|
|
Treatment of metabolic acidosis
|
1. identify/treat underlying cause
2. HCO3 replacement for severe academia (pH<7.1) esp w/ respiratory fatigue/heme instability, or for non-anion gap conditions. Be careful of overshoot metabolic alkalosis 3. Calculate HCO3 deficit = .5 x Body weight (kg) x ([desired HCO3] – [measured HCO3]). For severe acidosis bring to 10-12 mEq/L, titrate slowly |
|
|
Respiratory compensation for metabolic acidosis
|
- [H+] α Pco2/[HCO3]
- as ↓[HCO3] respiration increases to ↓PCO2 to maintain [H+] - after compensation: Pco2 = 1.5x[HCO3] + 8; if Pco2 differs >2mmHg then coexisting respiratory disorder: ↑Pco2 →respiratory acidosis, ↓Pco2 →respiratory alkalosis |
|
|
Common causes of metabolic acidosis
|
Increased anion gap:
- diabetic ketoacidosis: severe academia (<7.15), hyperglycemia, ECFV depletion, K depletion - lactic acidosis: hypotension/hypoxemia, disease (sepsis, organ failure, malignancy), medications - alcoholic ketoacidosis: minimal caloric intake (hypoglycemia), ECFV depletion, acute pancreatitis, GI bleed, Phos/Mg depletion, positive urine ketones - uremic acidosis: GFR <20%, sulfate/phosphate/anion retention - salicylate intoxication: initially respiratory alkalosis, then accumulation of organic acids from metabolic interference - ethylene glycol intoxication: acute CNS dysfunction, acute renal failure, calcium oxalate crystals in urine - methanol intoxication: optic neuritis (blindness from formaldehyde), pancreatitis - paraldehyde intoxication Normal anion gap: - mild/moderate renal failure: renal ammoniagenesis impairs H+ excretion, possible hyperkalemia - GI loss: acute secretory diarrhea, hypokalemia - Type I RTA: urine pH >5.3, hypokalemia (K wasting), possible calcium phosphate stones and nephrocalcinosis - Type II RTA: often assoc w/ Fanconi syndrome - Type IV RTA: aldosterone deficiency/insensitivity - Dilutional acidosis: rapid volume expansion (critical care) - treatment of diabetic ketoacidosis: ketones lost in urine |
|
|
Lactic acidosis types
|
L-lactic acidosis: most common form—pyruvate converted to L-lactate instead of Krebs cycle
Type A (Decreased tissue oxygenation): altered redox state, increased metabolic rate (sepsis, seizure, exercise, shivering, cancer, hypoglycemia, thiamine deficiency), decreased O2 delivery (hypotension/shock, severe hypoxemia, sepsis, CO toxicity, Pheo), hereditary metabolic defects (mitochondrial myopathies) Type B (decreased destruction/excretion): liver disease, ethanol, renal failure, drugs (metformin, salycilate abuse) D-Lactic acidosis: rare disease of short-bowel following bariatric procedure. Episodes of neurologic dysfunction induced by high D-lactic acid produced by fermentation of carbs by anaerobic gut bacteria |
|
|
Diabetic ketoacidosis
|
- occurs in severe, uncontrolled diabetes insulin resistance/insufficiency prevents cellular uptake of glucose, so ketones are produced for energy by incomplete oxidation of FAs and protein metabolism
- serum ketones are generally strongly positive and ionize inducing metabolic acidosis with anion gap Presentation: high anion gap metabolic acidosis, severe academia (pH <7.15), hyperglycemia, ECFV depletion, K depletion (despite normal or elevated serum levels), tachypnea, polyuria, polydipsia. - urine dipstick usually positive for ketones (does not detect beta-hydroxybutyrate, up to 75% of produced ketones) **alcoholic ketoacidosis: same mechanism, just chronic low carb intake lowers insulin levels and reduces gluconeogenesis |
|
|
Metabolic Acidosis
|
= decrease in plasma pH due to either increase in acid or decrease in HCO3
Acid etiologies (will increase anion gap, except tubular acidosis): increased endogenous hydrogen production (ketoacidosis, lactic acidosis, salicylate intoxication), toxic ingestion (methanol, ethylene glycol, paraldehyde), decreased renal excretion of hydrogen (uremic acidosis (chronic renal failure—anion retention), distal (type 1) tubular acidosis (normal AG)) HCO3 etiologies (normal anion gap): increased renal excretion (proximal (type II) tubular acidosis, mild/moderate renal failure), gastrointestinal loss in diarrhea High anion gap: addition of hydrogen plus unmeasured anion from either production of endogenous acids or addition of exogenous substances (toxins). H+ is buffered by HCO3 (lowering [ ]), while excess anion increases anion gap |
|
|
Central and nephrogenic diabetes insipidus
|
= syndrome of inability to concentrate urine due to poor production or response to ADH. Results in hypernatremia, extreme thirst and (dilute) polyuria (not reducible by fluid restriction—as polydipsia would)
- results from inability of the distal nephron to reabsorb water (rather than osmotic diuresis) - Central (poor ADH production/secretion): head trauma, post-neurosurgical (esp. pituitary), neoplastic, sarcoidosis, histiocytosis, meningitis/encephalitis, idiopathic. Treatable by administration of exogenous ADH - Nephrogenic (inadequate response to ADH): genetic (mutated V2 receptor, or AQP2 channel), electrolyte disorders (hypercalcemia, hypokalemia), drugs, recovery from acute renal failure, post-urinary obstruction, chronic renal disease (papillary necrosis, sickle cell). Not responsive to exogenous ADH |
|
|
Syndrome of Inappropriate ADH
|
= syndrome/symptoms associated with severe hyponatremia with normal volume status and normally concentrated urine (so not suffering from water loss, and not concentrating urine to correct hyponatremia). Usually normal renal, adrenal, thyroid function. Due to inappropriate production or response to ADH
4 mechanisms: increase in pituitary ADH secretion, ectopic (tumor) ADH production, ADH-like effect from exogenous substances, potentiation of ADH affects by drugs Etiologies: CNS disease, Pulm disease, Neoplasia, Post-operative state, Nausea, Drugs |
|
|
Hypernatremia
|
= [Na] <45mEq/L, generally results from loss of water and failure to replace it
- severe cases lead to cell shrinking causing lethargy, coma, intracranial bleeding (brain shrinks and hangs from veins) Etiologies: extra-renal water loss: insensible losses (fever, tachycardia, mechanical ventilation), sweat losses, GI losses (osmotic/infectious diarrhea). Renal water loss: osmotic diuresis (glucose, urea, mannitol), central diabetes insipidus (inadequate ADH), nephrogenic diabetes insipidus (inadequate renal response to ADH). Iatrogenic: administration of hypertonic solution |
|
|
Hyponatremia (with hypotonicity)
|
= [Na]< 135mEq/L
- rapid progression may result in brain swelling leading to lethargy, coma and seizures - results from impaired renal water excretion or Na reabsorption. Requires impaired renal water excretion Plus continued water intake - etiologies: impaired GFR (renal failure), ECF depletion (vomiting w/ continued water ingestion, secretory diarrhea), edema (CHF, cirrhosis, nephrotic syndrome), thiazide diruetics, SIADH, endocrine abnormalities (hypothyroidism or adrenal insufficiency/insensitivity), decreased solute + high water intake (tea& toast diet, beer potomania, primary polydipsia) - Normally low osmolality will trigger pituitary stop secreting ADH to increase water excretion and RAAS to increase Na retention, but these mechanisms are not effective to correct the problem - Tx: immediate water restriction until underlying etiology is understood and corrected (speed of Na replacement relative to speed of onset/symptoms) |
|
|
Hyponatremia with hypertonicity
|
= rare case of hyponatremia, usually associated with severe hyperglycemia (in uncontrolled DM) or administration of hypertonic mannitol
- [Na] is low but serum osmolatity and tonicity will be high - high glucose/mannitol load draws water from intracellular spaces diluting Na - [Na] falls approx. 1.6mEq/L for every increase in 100mg/dL glucose above 100mg/dL |
|
|
Pseudohyponatremia
|
= [Na] is low but measured ECF osmolality and tonicity are normal (calculated osmolality is low b/c of artificial low sodium)
- low sodium is an artifact due to accumulation of other plasma solutes - 2 main conditions: severe hypertriglyceridemia (1000’smg/dL) or severe hyperproteinemia (>10g/dL) - the patient should not be symptomatic and no treatment is required |
|
|
Plasma osmolality calculation
|
Osmolality = 2 x [Na] + glucose]/18 + [BUN]/2.8
Osmolar gap = Osm (measured) – Osm (calc); normally around 10, if greater unmeasured anions present in serum. Occurs in methyl alcohol and ethylene glycol ingestion |
|
|
Na transporters
|
NHE: Na/H antiporter w/ multiple isoforms. Most are basolateral and amiloride sensitive active in distal nephron and S3 of proximal tubule. Apical NHE in proximal tubule w/ apical H-ATPase and basolateral Na/HCO3 lead to net NaCL, HCO3 absorption
NKCC: thick ascending limb, target for loop diuretics, luminal K channels recycle K to run transporter so K is not limiting NCC: thiazide sensitive Na/CL cotransporter, active in distal CT. Apical so basal NaK ATPase then pumps Na out (Cl is through passive channels). Loss of function – Gittelman syndrome ENaC: aldosterone sensitive channel, permeable to H & Na, active in collecting ducts, coupled with basal Na/K ATPase. Can be blocked by triamterene, amiloride, or ANP H+ATPase (V-ATPase): essential for bicarb homeostasis, defect can cause tubular acidosis |
|
|
Cl reabsorption in the proximal tubule
|
- mainly passive along the PT, through tight junctions between cells
- In the early PT a gradient is established via Na cotransport of solutes and bicarb pulling water in and concentrating the chloride. By the late PT there is a positive gradient for reabsorption - ion countertransporters in the luminal membrane also contribute |
|
|
Salt/Water reabsorption in different nephron segments
|
Proximal tubule: 65% Na/H2O reabsorbed to the same extent, so [Na] is the same via Na/K ATPase (and various Na-X transporters) and passive water diffusion “iso-osmotic reabsorption”
Loop of Henle: 25% of Na (ascending, NKCC), 10% of water (descending), allows for creation of medullary gradient DCT/CD: 10% Na/Cl reabsorbed actively (NaCL in DT, ENaC in CD) and hormonally (aldosterone, allows for concentration), 5-24% water reabsorbed via ADH stimulation of aquaporins in CD |
|
|
Renal handling of weak acids and bases
|
- in neutral forms (non-ionized) organic acids/bases are more permeable to lipid membranes and can diffuse in/out of the lumen down concentration gradients. Ionized forms are essentially trapped in the lumen. pH therefore determines the relative concentration of acids/bases available for diffusion
- normally tubular lumen is acidified relative to plasma (US 5-6), so more weak acid will be neutral and permeable (less excreted) which bases are ionized and excreted - Carbonic anhydrase converts bicarb + H to water/CO2 so that it can diffuse and conserve bicarb - drug excretion/retention can be manipulated by changing the pH of the urine to retain or excrete it. |
|
|
Renal secretion of organic cations
|
- organic cations not bound to protein can undergo filtration and secretion (creatinine), while bound ones are secreted
- the proximal tubule has basolateral active transport systems that are relatively non-specific, and manifest Tm limitation Ex: acetylcholine, creatinine, dopamine, epinephrine, histamine, serotonin, NE, thiamine, and various drugs (atropine, cimetidine, morphine, procaine, quinine).9 |
|
|
Renal secretion of p-aminohippurate (PAH)
|
- PAH is an organic anion used to measure renal plasma flow (usually underestimates by 10%)
- it is completely filtered by the nephron (but only 20-30% by the glomerulus). Then it is actively secreted into the basolateral membranes of the proximal tubule, then passively diffuses into the lumen - PAH competes w/ many other organic anions for transport via the same basal transporter (low specificity) - Is not reabsorbed at all, so clearance can estimate renal flow |
|
|
Renal Handling of filtered proteins and small peptides
|
- little protein is filtered due to charge and size restriction ins the glomerulus
- Tm is low, and can be easily saturated if large quantities are filtered (glomerular disease) causing proteinuria - mechanism: binding of filtered protein to specific sites on the luminal membrane triggering endocytosis. Vacuoles the merge with lysosomes and proteins are degraded to amino acids which are secreted across the basolateral membrane into the peritubular capillaries - linear polypeptides (like ATII) are completely filterable and so are catabolized into amino acids in the kidney |
|
|
Renal handling of glucose
|
- reabsorption occurs mainly in the proximal tubule by secondary active transport
- SGLT1 (10%, proximal straight) and SGLT2 (90%, proximal convoluted) cotransport Na with glucose (as well as some passive glucose channels) reabsorb 100% of glucose - Tm for glucose is 375mg/min, plasma glucose above this as associated with proportional increase in glucose excretion (occurs in DM, Fanconi Syndrome) |
|
|
Renal handling of urea
|
- urea is highly diffusible in the kidney and so is gradient limited
- 50% absorbed in the proximal tubule, but more water is absorbed so it is concentrated. - it is passively secreted in the loop of Henle, so significant amounts reach the distal nephron - Little reabsorption occurs in the distal tubule or proximal collecting duct. The distal collecting duct has high permeability to urea so significant amounts are reabsorbed along with water, contributing to the medullary concentration gradient for countercurrent multiplication |
|
|
Different diffusion pathways
|
Transcellullar: occurs in regions of “tight” epithelia because the combined resistance to diffusion of ions over the apical and basal membranes is less than that of the TJs and intercellular spaces. Impermeable to large molecules
Paracellular: occurs through regions of “leaky” epithelia, where ions an small molecules pass through looser tight junctions directly into the intracellular space. Impermeable to large molecules |
|
|
Tubular maximum vs. gradient limited transport
|
Tubular maximum:
- reabsorption limited by the saturation of high-affinity transporters (specific to them, but may transport a few substances--competition), so increase in solute concentration does not increase speed. May be actively resorbed or symported (usually w/ sodium). Usually limit is well above normal concentrations so solutes may be completely reabsorbed - Ex: glucose (prox tub), amino acids, weak acids, proteins, organic molecules, nucleic acids, TCA intermediates, vitamins, lactate, acetoacetate, β-hydroxybutyrate Gradient-limited: - reabsorption limited by passive diffusion, not saturation of transporters (transporters used but not rate-limiting). Ex: Na leaks back faster than transporters absorb if gradient is favorable - characterized by degree of back-leak through tight junctions (occurs when interstitial concentration builds reducing osmotic gradient) - Ex: Na, Cl, H2O, HCO3, most electrolytes and other tight junction-permeable molecules (urea) |
|
|
Electrolyte abnormalities associated with thiazide or loop diuretics
|
Loop diuretics act by inhibiting the NKCC2 transporters in the LoH preventing Na/Cl reabsportion (main function of LoH) causing volume loss. This is usually compensated by activation of RAAS system
Thiazides inhibit Na/CL cotransporter in the distal tubule, also causing volume loss and RAAS activation BOTH result in hypokalemia and metabolic acidosis via action of aldosterone to absorb Na in the distal tubule at the expense of K and bicarb Thiazides cause hypocalcuria/hypercalcemia due to excess calcium reabsorption, while Loops do the opposite (hypercalcuria may lead to kidney stones) Gittleman syndrome causes congenital absence of Na/Cl cotransporters to patients present as if on excess thiazides |
|
|
Fanconi Syndrome
|
- autosomal recessive disorder causing global dysfunction of the proximal renal tubules (so they don’t absorb anything: glucose, water, bicarb, ions, phosphorus, amino acids, urea, etc)
- most commonly caused by cysteinuria, which results in massive loss of nephrons - often manifests at a young age with: metabolic acidosis (bicarb wasting), hypophosphatemia and Rickets (phosphate wasting), glycosuria (poor glucose reabsorption, but not DM), low plasma uric acid, amino aciduria |
|
|
Substances reabsorbed in the proximal tubule
|
Bicarbonate: major buffer, altered reabsorption leads to metabolic acidosis/alkalosis, can be blocked by acetazolamide
Phosphorus: excess loss causes osteomalacia (adults) or Rickets (children Glucose: should all be reabsorbed, if not pathologic – DM, others Water: 65% reabsorbed with the solutes Urea: 50% Amino acids: can be diagnostic Uric acid: can be diagnostic |
|
|
Etiologies of aldosterone pathogenesis
|
Low Aldosterone leads to hypotension (Na/H2O wasting), hyperkalemia (K retention), metabolic acidosis (H retention)
High Aldosterone leads to hypertension (Na/H2O retention), hypokalemia (K wasting), metabolic alkalosis (H wasting) Diseases: - Adrenal adenoma: aldosterone producing tumor—excess aldosterone - Liddle syndrome: constitutively open ENaC channels—effective excess aldosterone (though actually low in plasma) - 21-Hydroxylase Deficiency: loss of enzyme required for aldosterone synthesis so unresponsive to ATII (results in adrenal hyperplasia) – aldosterone deficiency - Pseudohypoaldosteronism Type 1: genetic absence of ENaC channels – effective aldosterone deficiency (high serum levels) |
|
|
Aldosterone activity in the kidney
|
- acts on the distal collecting duct to open apical ENaC channels (allowing Na influx into epithelial cells from the lumen) and upregulate basal Na/K ATPases (increasing Na reabsorption and K excretion)
- Net movement of Na leave a negative charge drawing loss of K and H ions, and promotes water intake (though that’s mainly mediated by ADH) |
|
|
Autoregulation of RBF
|
= the process of controlling RBF to keep a constant ΔPuf and GFR. This occurs between BP of 80-200mmHg
- intrinsic function of the kidney conducted by balance of the smooth muscle tone in the afferent and efferent arterioles - guided by flow equation: Q = ΔP/R (so increase in P will result in increase of resistance) - RBF is inversely related to the sum of efferent/afferent resistance, which GFR is controlled by the ratio of the two - this function is mediated by tubuloglomerular feedback: changes in [salt] in the filtrate is detected by the macula densa causing alteration of arteriole resistance - another regulatory mechanism of GFR is for the podocytes and mesangial cells to alter glomerular surface area |
|
|
Starling forces
|
Starling forces determine ultrafiltration: ΔPuf = (Pgc + πbc) – (Pbc + πgc)
- For filtration: hydraulic pressure in the capillary, Oncotic pressure in Bowman’s capsule - counter filtration: hydraulic pressure in the bowman’s capsule, oncotic pressure in the capillary (significant because of protein load, increases toward the efferent ateriole so net filtration decreases) Renal disease will change the number and viability of nephrons rather than the starling forces within the nephron |
|
|
Normal renal blood flow parameters
|
Normal RBF = 1.1L/min, determined by renal artery pressure and SM contraction of the arterioles in the cortex
Normal renal plasma flow = 0.55x1.1L/min = 605mL/min Normal GFR = 125 mL/ min Filtration fraction = GFR/RPF = 20% (normally), the remaining 80% is diverted to the peritubular capillaries |
|
|
Determinants of GFR
|
GFR = hydraulic permeability X surface area X net filtration pressure = Kf X ΔPuf (perm coefficient X ultrafiltration pressure)
- permeability is only altered by pathology, surface area can be altered by podocytes and mesangial cells - Puf is always positive due to starling forces, but varies proportionally with RBF when BP <80 or >200 (due to autoregulation) - glomerular capillaries filter more than peripheral capillaries despite less surface area because the pressure (Puf) are higher |
|
|
Fiterability of plasma solutes through the glomerulus
|
Filtration barrier:
- fenestrated epithelium: allows passage of anything <70kDa (most ions, sugars, amino acids, hormones), excluding large proteins (albumin), cells, and hydrophobic proteins (thyroid and steroid) bound to larger transport proteins - basement membrane (protein mesh with heparin sulfate): negatively charged so excludes larger negatively charged compounds (proteins) - slit diaphragm (between processes of podocytes): does not significantly contribute to filtrations (but help prevent plasma protein leakage) |
|
|
Urinary casts
|
- formed by precipitation of Tamm-Horsfall protein (THP) in distal tubule and collecting ducts, giving tubular appearance
- most likely to precipitate in low-flow, low pH, or concentrated salt conditions. - trap structures in the tubules and then dislodged into the urine Types: - hyaline (faint, almost invisible): physiologic in dehydration state, also suggest CKD - Fine, granular: granular material is degradation of cells or proteins, usually in kidney injury - waxy or broad: have wider lumen, clear; signify atrophic tubules in ongoing CKD or in malignant HTN - RBC: visible brown cells; hallmark of glomerularnephritis - Muddy brown: brown due to mitochondria; characteristic of tubular ischemia causing acute tubular necrosis (often associated septic shock, renal failure) - WBC: white cell aggregate; mostly pyelonephritis, interstitial nephritis, or glomerulonephritis - Fatty: visible under polarized light; from fatty degeneration of the renal epithelium, typically glomerulonephritis |
|
|
Crystals visible in urinalysis
|
Calcium oxalate: needle shaped, envelope or dumbbell shaped, major component of kidney stones.
Uric acid: yellow to orange-brown, different shapes (diamond, barrel) depending on urine pH, light up under polarized light, typically in acidic pH Calcium phosphate: colorless, coffin-lid appearance, may look like uric acid but appear at alkaline pH and UTI, rare Cystine: hexagonal crystals, present in cystinuria but also common |
|
|
Findings on urinalysis
|
Appearance: should be clear not cloudy
Color: orange (contrast dye, drugs-rifampin), pink/red (RBCs, beets, food coloring), white (pus, WBCs) Odor: maple syrup (MSU disease), musty (PKU) Specific gravity (normal 1-1.03): 1.000 is dilute (water), >1.03 is highly concentrated (hypertonic) pH: depends on diet (normal US 5-6); increased (urea splitting organisms), decreased (bacterial metabolizing glucose to acids) Hemoglobin: hematuria (can’t distinguish from myoglobin though) Glucose (max 180mg/dL): >180 w/ high serum (diabetes), w/ normal serum (Fanconi) Protein (normal 80mg/d, varies w/ age, activity, disease): high (over 24hrs) glomerulonephritis Ketones (acetoacetate, acetone): does not test for beta-OH-butyrate (sign of DM ketoacidosis) Bilirubin: nor normal for urine (only conjugated may be present) Urobilinogen: increased w/ jaundice (breakdown from bilirubin) Leukocyte esterase: positive if >5 WBC/HPF, usually UTI (also interstitial nephritis) Nitrites: mostly from nitrate by bacterial transformation (Pseudomonas, proteus) RBCs: any source in urinary tract (identify w/ history), early urination (urethra) late urination (bladder) continuous (kidney upper urinary). Absence of proteinuria usually r/o glomerular source. Different mortphologies: normal, or acanthocytes or crenated (distorted) suggestion glomerular disease Crystals: calcium oxalate, uric acid, calcium phosphate, cysteine, cholesterol Microorganisms: cocci, bacilli, yeast w/ pseudohyphae or budding, trichomonas (suggests STI, immediate treatment required) Fecal matter: abnormal fistula or external contamination Ammonium biurate: advanced liver disease Mucous: (rare)UTIs, STDs, ulcerative colitis, other intestinal problems Spermatozoa: w/in 24hrs of ejaculation |
|
|
Reabsorption in different segments of the nephron
|
Proximal tubule: 2/3 of water, sodium and chloride; 90% if bicarb, 100% of glucose and amino acids, some of other electrolytes (K, Phos, Ca). Secretion of waste products (urate, creatine, drugs)
Descending LoH: passive reabsorption of water (allows for concentration of urine) Ascending LoH: 15-25% of electrolytes (Na, K, Cl). Allows for dilution of urine and establishment of hypertonic medullary interstitium (more salt than water absorbed) Distal tubule and connecting tubule: 5% of additional salt and water (tight epithelia), mostly through apical Na/Cl cotransport and basal Na/K ATPases making urine hyper tonic Collecting duct: 5% of Na (responsive to aldosterone), K excretion (also aldosterone), water reabsorption (ADH responsive), urea absorption/excretion |
|
|
Evolutionary development of the nephron
|
1. (Saltwater fish) Development of the proximal tubule to filter and retain nutrients (amino acids, glucose) and excrete isotonic urine
2. (Freshwater fish) Development of proximal straight and distal tubule to conserve electrolytes and produce dilute urine 3. (Land animals) Development of hormonal control over water reabsorption in the collecting tubule and ability to concentrate urine in the loop of Henle. |
|
|
7 major function of the kidney
|
1. Regulation of water and electrolytes (input matches output at a steady state)
2. Regulation of arterial blood pressure 3. Excretion of metabolic waste 4. Excretion of foreign substances 5. Regulation of red blood cell production 6. Regulation of the active form of vitamin D 7. Gluconeogenesis (only important during significant fasting) |
|
|
Specialized cells of the collecting duct
|
Principal cells: mediates Na/K balance via apical Na and K channels. Aldosterone determines expression of Na channels and Na/K ATPase pumps. ADH determines expression of aquaporins
Dark/intercalated cells: participate in acid-base homeostasis by secreting and resorbing acid and bicarb with alpha and beta cell types |
|
|
Renin-angiotensin-aldosterone system
|
Secretion of renin into the blood stream by JG cells occurs in response to low arterial BP (baroreceptors), low NaCl in filtrate (macula densa cells), or sympathetic stimulation (beta1 receptors).
Renin catalyzes formation of ATI from angiotensinogen (produced in liver). ACE (found on vascular endothelial cells) then converts ATI to highly active ATII, which travels to kidneys to increase sympathetic activity, increase Na/Cl reabsorption, arteriolar vasoconstriction, and ADH secretion from the pituitary gland and aldosterone secretion from the adrenal glands. Net result is increase salt and water retention, increasing effective circulating volume (an increased JG perfusion) |
|
|
Juxtaglomerular apparatus
|
- complex of cells found between the vascular pole of the glomerulus and the returning distal convoluted tubule
JG cells (granular cells: specialized smooth muscle cells in the walls of the arterioles (mostly afferent) that synthesize, store, and secrete renin. Act as interrenal baroreceptors (monitor BP/vascular volume in the last portion of the afferent arteriole, vary renin secretion inversely) Macula densa cells: increased luminal flow results in increased Na/Cl accumulation in the cells, triggering increased production of vasoconstrictors and feed back to glomerular cells to inhibit renin release Extraglomerular mesangial cells: light staining, specific function not understood (help with other cellular functions |
|
|
Epithelial cells of the nephron
|
- renal corpuscle: podocytes at the vascular pole transition to squamous epithelium, then a the urinary pole transition again
- Proximal tubule: cuboidal or columnar cells, allows for extensive reabsorption of salts and proteins. Cells have lots of mitochondria, elaborate basolateral folds (Na/K ATPases), apical brush border (do active endocytosis) - LoH: squamous epithelium specialized for H2O permeability. Descending has brush border, ascending does not. Contain lots of mitochondria for active molecular transport. Looks like vessel w/o RBCs - Distal tubule: simple columnar epithelia, permeable only by ADH stimulation, no brush border but occasional microvillus, lots of basolateral folds and mitochondria. Shorter so will see fewer in cross-section (also smaller and more regular shaped). Includes macula densa cells (small, jumbled nuclei, dense patchwork appearance) - Collecting duct: lacks elaborate basolateral folds - Renal pelvis, calyces, and ureter: stratified epithelium - Bladder: transitional epithelium (dome shaped apical surfaces, often paired nuclei) |
|
|
Mesangial Cells
|
= modified smooth muscle cells that lie inbetween the capillaries and the glomerulus
- control the caliber of the capillaries by contractile activities, pull on basement membrane, adjust local blood flow though capillaries - phagocytose proteins caught in the basement membrane |
|
|
Diagnostic process for glomerulonephritis
|
For suspected renal dysfunction:
- examine extra-organ manifestations (fever, fatigue, arthralgias, rash, hair/weight loss, edema) - establish baseline kidney function: labs (creatinine, GFR) - blood tests for specific etiologies: ANA (SLE), complement activation (SLE, SBE, post-strep, MPGN), ANCA (vasculitis), anti-GBM (Goodpasture’s), ASO (strep), cyroglobulins, hepatitis - complement levels: LOW (SLE, SBE, Shunt nephritis, cryoglobulinemia, acute post-strep, membranoproliferative GN), NORMAL (IgA nephropathy, PAN, Wegener’s, Goodpasture’s, hypersensitivity vasculitis, idiopathic rapidly progressing GN) |
|
|
Presentations of glomerulonephritis
|
- proteinuria, due to: diseased glomeruli (damaged filtration barrier), increased serum protein exceeds tubular maximum (heme malignancy), low reabsorption in the proximal tubule (heavy metal poisoning, Fanconi syndrome)
- edema formation: increased proteinuria leads to hypoalbuminemia and low plasma oncotic pressure, leading to interstitial fluid collection. Also activates RAAS to increase Na retention (increases fluid). Na retention may be primary mechanism (increases hydrostatic pressure) - Complications: hyperlipidemia (increased synthesis, decreased lipase activity), immune deficiency (lose antibodies, PMNs), hypercoagulability (due to protein loss/coag regulators) |
|
|
Diffuse proliferative glomerulonephritis
|
- a nephritic syndrome due to systemic inflammatory diseases (SLE, RA) and vasculitis syndromes (Wegener’s). Characterized by >50% of glomeruli showing proliferation in mesangial, epithelial, endothelial, and inflammatory cells.
Pathogenesis: mostly due to deposition of immune complexes in the mesangium, basement membrane, or subendothelial or subepithelial spaces. Results in obliteration of capillary loops and sclerosis |
|
|
Post-streptococcal glomerulonephritis
|
= nephritic syndrome associated with infection by Group A β-hemolytic streptocci (strep throat or impetigo). Usually presents 1-4wks post infection, more common in children, self-limited
Presentation: 1-2wks post infection, abrupt onset of periorbital edema (mild proteinuria <1g/d), malaise, fever, nausea, oligouria, and gross hematuria (smoky/coca-cola colored; RBC casts). Labs show: ↑ASO, hypocomplementemia, +/- cyroglobulins Pathogenesis: type III hypersensitivity reaction to strep antigen causing immune complex production, sub-epithelial IC deposition + in situ formation. Classical complement activation and cross-reaction w/glomerular antigens mediating GMB destruction Histo: -LM: diffuse proliferative GN (enlarged & hypercellular), neutrophilic infiltrate, endocapillary proliferation (histocytes, lymphocytes can occlude capillary loops), lumpy-bumpy appearance - IF: granular/starry sky appearance due to IgG, IgM and C3 deposition - EM: subepithelial humps, no spike formation |
|
|
Membranous nephropathy
|
- second most common nephrotic syndrome in adults, slowly progressive disease beginning btwn 30-50 characterized by a thick glomerular basement membrane, proteinuria and elevated creatinine
- 85% are primary/idiopathic; 15% secondary to autoimmune disease (SLE), infections (syphilis, malaria, Hep B), drugs (NSAIDs), malignancy (carcinoma’s, Hodgekin’s) Histo: - LM: increased mesangial matrix w/ thickened capillary loops (PAS and silver—also shows lucent areas of spike formation) - IF: granular IgG along capillary loops - EM: thick basement membrane with subepithelial dense deposits with spike/dome changes (indicating chronicity) Treatment: ACE inhibitors, steroids, cyclophosphamide if refractory Prognosis: 60% have persistent proteinuria, 40% progress to renal insufficiency, 10-30% have partial or complete remission of proteinuria |
|
|
Focal segmental glomerulosclerosis (FSGS)
|
- most common nephrotic syndrome in adults characterized by focal (<50%) and segmental sclerosis of the glomeruli
- present with hematuria, proteinuria, hypertension. Tend to have poor response to steroids (progressive scaring and loss of function), >50% progress to renal failure in 10yrs. - Mostly idiopathic, can be secondary from HIV (collapsing variant), toxins (heroin, pamidronate), any disease causing altered renal flow (hypertension, sickle cell anemia) Histo: - LM: focal (need enough biopsy to r/o MCD), segmental increase in mesangial matrix with sclerotic segments of glomerulus (contain glassy, proteinacious hyaline matrix) - IF: none (likely not immune mediated) - EM: diffuse foot process effacement, no dark deposit |
|
|
Minimal change nephropathy
|
= most common cause of nephrotic syndrome in children causing proteinuria and edema but preserved renal funtion. Primary glomerular disease.
- Idiopathic damage to podocytes, typically insidious onset (otherwise healthy child), 95% respond to steroids w/ v. good prognosis (5% progress to CKD) - Characterized by lack of pathology on LM or IF (likely not autoimmune). ONLY evidence is on EM: diffuse foot process effacement (this + proteinuria + preserved renal function is diagnostic in the absence of other causes) |
|
|
Nephritic syndrome
|
= collection of signs associated with disorders of the glomerulus characterized by podocytes that permit filtration of protein (proteinuria) and cells (hematuria)
Signs: - hematuria (macro/micro, +/- RBC casts)—main issue - acute renal failure: not always but usually some degree of azotemia (↑BUN) and/or oliguria (↓urination) or ↑CrCl/↓GFR - hypertension - proteinuria and edema (not as severe as nephritic syndrome, <3.5g/d) - Urinalysis: hematuria, RBC casts, proteinuria Etiologies: acute post-streptococcal GN, rapidly progressive GN (cresentic GN), diffuse proliferative GN, IgA nephropathy (Berger’s), Alport’s syndrome |
|
|
Nephrotic syndrome
|
= collection of signs associated with disorders affecting the glomerulus characterized by pores in the podocytes that permit protein filtration but not cells
Signs: - massive proteinuria (>3.5 g/d), but no hematuria - Edema (significant compared to nephritic disease) - hypoalbuminemia (from proteinuria) - hyperlipidemia - relatively preserved renal function at presentation (Cr may be normal) - Urinalysis: severe proteinuria, oval fat bodies, minimal or microscopic hematuria Etiologies: membranous glomerulonephritis, minimal change disease, amyloidosis, diabetic glomerulonephropathy, focal segmental glomerulosclerosis, membranoproliferative glomerulonephriti |
|
|
Etiologies of Hematuria
|
Renal:
- parenchymal: malignancy (eg. Renal cell carcinoma), polycystic kidney disease, analgesic nephropathy, papillary necrosis, sickle cell nephropathy, vascular (aneurysm, renal infarction, renal vein thrombosis, malformations), crystaluria - glomerular: primary renal disease (glomerulonephritis), multisystem disease (collagen vascular diseases, SLE, Goodpasture’s disease, systemic vasculitis, hemolytic uremic syndrome), heredofamilial (Alport’s syndrome, thin glomerular basement membrane disease Extra-renal: ureters, urethra, prostate, bladder, etc - Hematological: bleeding diathesis, anticoagulation (warfarin, heparin), disseminated intravascular coagulation - Urinary tract: urolithiasis, UTIs (cystitis, prostatitis), malignancies (urethral, prostate, bladder, ureter, renal pelvis),benign prostatic hypertrophy, drugs (hemorrhagic cystitis caused by cyclophosphamide, warfarin) |
|
|
Fuhrman criteria for grading malignant renal neoplasms
|

- Grade I - small, round, uniform nuclei (10 microns), look like lymphocytes [ <5% of tumors]
- Grade II - slightly irregular nuclei, see nucleoli at 40x only, nuclear diameter 15 microns [40% of tumors] - Grade III - nuclei very irregular, see nucleoli at 10x, nuclear diameter 20 microns, [30-40% of tumors] - Grade IV- mitoses, bizarre, multilobated, pleomorphic cells plus grade 3 features, macronucleoli [<20% of tumors] |
|

|
Wilm’s Tumor/Nephroblastoma
= most common renal tumor in children (1/8000), 90% <6yrs, M=F Presentation: large flank mass +/- hematuria. May have coughing (lung metastases) or traumatic rupture - Triphasic: contains blastema (un-differentiated glomerular cells), epithelium (gland forming), stroma (mostly spindle cells) - Prognosis: very aggressive with rapid progression, often fatal. Treat with Nephrectomy and/or chemo Associated with: - WAGR syndrome: Wilm’s tumor, Aniridia (no iris), Genitourinary malformation, mental-motor Retardation - Denys-Drash Syndrome: gonadal dysgenesis (male pseudohermaphroditism), glomerulosclerosis, Wilm’s tumor - Beckwith-Wiedemann syndrome: exophthalmos (bug eyes), macroglossia, gigantism, also hemihypertrophy |
|

|
Urothelial Carcinoma
= malignant tumor urinary tract. May arise in the renal calyces, renal pelvis, ureters and bladder - primarily affects adults (70% M, mean 70yrs), 7% of all primary renal carcinomas - present with painless hematuria (tumor erodes urothelial lining exposing vessels) - 40-50% of patients with kidney tumor will have pre/co-existing bladder tumor - Associated with (PeeSAC): phenacetin, smoking, aniline dyes, cyclophosphamide. Also horseshoe kidney - treatment: neuphroureterectomy (have to take out kidney and part of the ureter) |
|
|
Papillary Renal cell carcinoma
= malignant renal tumors arising from either proximal or distal tubules - Epi: 10-20% of all adult renal carcinomas, M:F = 3:1 - associated with trisomy 7, trisomy 17, Y chromosome loss, rare hereditary forms Gross: large, hemorrhagic, may be necrotic Histo: large foamy histocytes, calcifications (in more malignant tumors), |
|

|
Clear cell Renal Cell carcinoma
= malignant renal tumor arising from proximal convoluted tubules - Epi: 70% of adult renal epithelial tumors, usually onset >50yrs, M:F = 2:1, 1% bilateral - increased incidence with smoking, obesity (esp. women), chronic HTN, tuberous sclerosis, von Hippel-Lindau (50%, deletion f VHL gene 3p25) - Presentation: hematuria, palpable mass, 2° polycythemia, flank pain, fever, weight loss. Maybe asymptomatic w/ incidental finding - Spread: along IVC then hematogenous to lungs , lymph, liver, bone, adrenals, collateral kidney, brain, etc - Treatment: total or partial nephrectomy, cryblastion - May be associated with paraneoplastic syndromes: ectopic EPO, ACTH, PTHrP, prolactin Histo: gross: yellow/fatty appearance; micro: round nuclei, pale cytoplasm (lipid, glycogen inclusions washed out), highly vascular (chicken wire appearance) |
|

|
Oncocytoma
= benign renal tumor - incidence: 4-7% of adult renal epithelial tumors, onset mostly >50 (but can have wide range), M:F = 2:1 - majority asymptomatic, may be multifocal and/or bilateral - Histo: brown lesion with characteristic central scar, uniform population of pink cells, loose fibrous stoma, island-like architecture |
|

|
Angiomyolipoma
= benign renal neoplasma composed of thick walled vessels, smooth muscle, and fat vesicles. - incidence: <1% of all renal tumors - Associated with tuberous sclerosis: these patients can havelarge, multifocal lesions - Lesions >4cm can cause spontaneous hemorrhage (present w/ severe back pain)due to increased vasculature and so should be resected (small ones are not at risk) - Histo: contain deposits of blood vessels, smooth muscle, adipocytes |
|

|
Papillary adenoma
= Benign cortical lesions, usually <5mm well circumscribed - incidence increases with age (and detection increases w/ imaging), associated with long term hemodialysis. Mulitple adenomas associated with papillary renal cell carcinoma - Histo: small lesions with tubulopapillary architecture (gland forming) |
|
|
Dense deposits
|

= collections/condensation of immune complexes. May be subendothelial (btwn endocytes and GVM), intermembranous (in GMB), subepithelial (btwn GBM and FPs), or mesangial (the lead to cell activation causing increased cellularity and matrix secretion).
|
|
|
Osmolarity vs. volume regulation
|
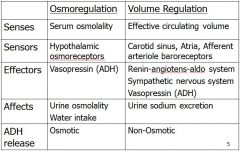
|
|
|
Rapidly progressing (crescentic) glomerular nephritis
|
= histologic pattern of glomerular disease associated with nephritic acute renal failure. Poor prognosis w/ renal function deteriorating within days/weeks
Pathogenesis: anti-NC1 antibodies (collagen IV domain) cause membrane rupture, fibrin deposition in the urinary space and activation of parietal epithelial cells. Types: limited to kidneys → Anti-GBM disease; also attacks alveolar membrane → Goodpasture’s syndrome (pulmonary-renal syndrome: respiratory distress/pulmonary hemorrhage + acute renal failure) Histo: - LM: fibrin/plasma protein (C3b)/cellular crescents in urinary space with monocytes/macrophages - IF: linear IgG deposition - EM: no dense deposits visible |
|
|
Membranoproliferative glomerulonephritis Type II
|
= “Dense deposit disease” disease associated with partial lipodystrophy (abnormal fatty deposits).
Pathogenesis: 70% have circulating C3 nephritic factor (C3NeF) which stabilizes C3 convertase and promotes alternate complement pathway activation. Patients have C3 hypocomplementemia (normal C1/4) and diminished Factor B and properdin (alternate pathway components). Histo: - LM: diffuse mesangial proliferation, endocapillary proliferation - IF: thick, linearized C3 deposition plus some mesangial deposits - EM: linear bands of dense deposits within the membrane, foot process effacement |
|
|
Membranoproliferative glomerulonephritis Type I
|
= glomerular disease with either nephritic (hematuric) or nephrotic presentation or combined. Poor prognosis (>50 develop CRF w/in 10 years), children (10-20% of all cases)>adults
Histo: -LM: diffuse mesangial proliferation, tram-tracking esp on silver (thickened capillary loops from mesangial interpositioning), +/- endocapillary proliferation - IF: granular C3 deposition along capillary loops - EM: subendothelial deposits, effaced and normal FPs |
|
|
Glomerular diseases with Nephrotic presentations
|
Focal segmental glomerular sclerosis: HIV
Membranous GN: SLE, drugs, tumors Minimal Change Disease Amyloidosis Membranoproliferative GN Diabetic glomerulonephropathy |
|
|
Glomerular diseases with Nephritic presentations
|
Acute post-streptococcal GN
Rapidly progressive (crescentic) GN: Goodpasture’s syndrome, Wegener’s granulomatosis, Microscopic polyangitis Diffuse proliferative GN: SLE, membranoproliferative GN IgA Nephropathy (Berger’s Disease): related to Henoch-Schonlein disease Alport Syndrome |
|
|
IgA nephropathy (Berger’s Disease)
|
= IgA mediated mesangiopathic disease with gradual, insidious progression (decades) to end-stage renal disease. Characterized by significant alteration in IgA glycosylation patterns in hinge region leading to aggregation/deposition (underglycosylation)
- no effective treatment; related to Henoch-Schonlein disease; most common GN in the developing world Presentation: nephritic--asymptomatic hematuria (gross or macroscopic) +/- proteinuria. Histo: - LM: normal capillary loops but increased mesangial matrix deposition and hypercellularity (>3 nuclei/stalk) - IF: IgA with mesangial pattern of deposition - EM: expanded mesangial matrix with dark deposits, normal FPs Theoretical mechanism: IC deposition stimulates proliferation but impairs collagen IV secretion resulting in GMB thinning/attenuation leading to rupture and hematuria |
|
|
Diabetic nephropathy
|
= renal damage due to chronic diabetes (I or II)
Mechanism: unclear, 3 theories - persistent high glucose results in non-enzymatic glycosylation of proteins resulting in advanced glycosylation end products (AGEs) which can damage tissue over time - high glucose activates PKC pathway, which in certain conditions generates excess pro-angiogenic factors (VEGF) leading to nephropathy - Disturbance in polyol pathway: excess glucose metabolized to sorbitol (polyol) then fructose which depletes antioxidant reserves predisposing the kidney to damage Clinical presentation: initially trace microalbuminuria (30-300mg/d) from early damage. Kidney increases GFR due to leakiness leading to enlargement. Increasing albuminuria as disease progresses, advanced disease may present w/ nephrotic syndrome Histo: - LM: GBM thickening (+/- change in renal function), diffuse mesangial sclerosis (proliferation and increased secretion, usually after >10yrs). Late disease: Kimmelstiel-Wilson nodules (15-30% of diabetics, nodular expansion of mesangial matrix), plasma protein inclusions (hyalinosis lesions), afferent & efferent hyaline arteriolosclerosis, scaring of the tubulointerstitium |
|
|
Causes of nephrotic syndrome
|
Primary glomerular diseases: 95% of pediatric cases, 60% of adults
- Minimal change disease - membranous nephropathy - focal segmental glomerulosclerosis Systemic diseases w/ renal manifestations: 5% of peds, 40% of adults - Diabetes mellitus - amyloidosis - SLE - Drugs (gold, penicillamine, heroin) - Infection (malaria, syphilis, hepatitis, HIV) - Malignancy (carcinoma, lymphoma, melanoma) - Bee sting allerg |
|
|
Pathophysiology of nephrotic syndrome
|
- Always begins with derangement the glomerular filtration barrier (ex: deposition of immune complex) resulting in increased permeability and loss of plasma proteins in the urine
- Profound proteinuria (esp. albumin, also immunglobulins, complement factors, anti-coagulant proteins) will result in losses greater than the liver can recoup, resulting in hypoalbuminemia, hypercoagulability, and hypocomplementemia. - Lipoprotein synthesis is upregulated, lipoprotein lipase activity is decreased resulting in hyperlipidemia and lipiduri |
|
|
In-center vs. home dialysis
|
Home:
- requires home care-giver in case of emergencies - 6x/week, each is shorter duration/volume, allows for more stable levels of blood components (avoids extremes) - dialysate flow is lower than blood flow, allowing more solute transfer for dialysate volume resulting in higher dialysate saturation - generally better quality of life: faster recovery from treatment, less complications due to more stable blood levels (can resolve calcium phosphate deposits, tend to have lower BP) Center: - 3x/week, larger volume/time per session - significant water requirements |
|
|
Complications of dialysis
|
- hypotension (pull too much fluid off the patient) due to: interdialytic weight gain, severe LVF, MI, pericardial tamponade, sepsis, hemohrrhage, arrhythmia, dialyzer reaction, hemolysis
- Muscle cramps due to: hypotension, below dry weight, low dialysate sodium (vasoconstriction) - Others: restless leg syndrome, nausea/vomiting, headache, disequilibrium syndrome (due to brain swelling/rapid ↓pressure—n/v restlessness, headache, seizures, obtundation, coma) - From machine set-up: kinks in the tube or fluid temperature can cause hemolysis, which can increase potassium |
|
|
Indications for dialysis therapy
|
Emergent (to avoid serious short-term side effects:
- hyperkalemia, acidosis, uremic pericarditis, uremic encephalopathy, volume overload, toxin exposure Long term (use albumin and nutritional status as survival indicatiors: - uremic syndrome, significantly reduced creatinine clearance (10-15 mL/min) |
|
|
Physiology of dialysis
|
- Diffusion: removes blood products by allowing them to travel down concentration gradients into the dialysis fluid (absent of blood products initially). Inhibited by membrane resistance and molecule size (large ones diffuse worse).
- Ultrafiltration: hydrostatic pressure forces solvent through the membrane, increasing the removal of poorly diffusible solutes (though tends to exclude large solutes). - Overall: two mechanisms provide good filtration of most blood components while retaining blood and proteins. Dialysis does not filter protein-bound compounds well though, which may be detrimental long term for the patient |
|
|
Types of Renal Replacement therapy
|
Hemodialysis: filters the blood through direct vascular access. Short term/high acuity patients (subclavian or femoral veins), long term patients (arteriovenous fistulas, shunts or grafts in the lower arm—required to speed flow to machine).
Peritoneal dialysis: uses the peritoneal membrane as the dialysis membrane—fluid is put into the peritoneal space blood components diffuse into it Transplant: ultimate replacement for defective kidney function |
|
|
Prostatic carcinoma
|

= androgen dependent adenocarcinoma arising primarily in the peripheral zone of the prostate
Epi: 2nd most common cancer in men and #2 cause of cancer death Risk factors: African America (Asian is least), age >50, familial incidence, high sat. fat diet (lycopene protective?) Path: glandular formation with large nucleoli (look like eyes) Presentation: initially asymptomatic, once large can cause prostatism (via urethal compression). Can invade causing systemic symptoms Spread: direct (seminal vesicles, base of the bladder), lymphatic (obdurator lymph nodes), hematogenous (vertebrae, pelvis, proximal femur). Osseus metastases are typically osteoblastic resulting in sclerotic structures (rather than destructive) Labs: ↑OSA, ↑PAP (prostate acid phosphatase), ↑ALP (osteoblastic metastasis) Gleason grading (1-10) based on architecture rather than nuclear atypia, average presentation is 6 Tx: prostatectomy w/ radiation, anti-androgen therapy (orchiectomy, estrogen, analogs of GnRH, flutamide (androgen receptor inhibitor) |
|
|
Nodular prostate hyperplasia
|

= BPH involving nodule formation, can result in prostatism
- Gross: hyperplastic +/- bladder hyperplasia. Circular nodules visible in cross-section, medial lobe can push up into the bladder, peripheral zone is compressed - Histo: circular nodules consisting of hyperplastic stromal and glands, which secrete alkaline, milky fluid for ejaculation |
|
|
Testicular cancer
|
Peak incidence age 15-34, most often involving germ cells, discovered by self-exam, highly curable
Risk factors: undescended testis, genetic conditions, HIV infection, atrophy, ?Fx, ?trauma Lymphatic spread: drains along spermatic cord to retroperitoneal nodes (not scrotum), less common to get bilaterally Penile cancer: rare, almost always in uncircumcised males, usually SCC, spread by local extension |
|
|
Peyronie’s Disease
|
= acquired, abnormal curvature/deformity of the penis during erection that interferes with sexual intercourse and often causes psychological stress to the patient
-10% of men by age 50-60, usually Caucasian - thought to be trauma related (in a predisposed individual) resulting in abnormal fibrosis/scar formation (dorsal tunica layer tears), may be associated w/ ED in later stages b/c of vein abnormalities |
|
|
Erectile dysfunction
|
= inability to achieve and/or maintain an erection sufficient for sexual function
Etiologies: vascular, neurologic, psychogenic, endocrine Normal: stimulation→ NO release→ guanylate cyclase activation→ ↑corporal cGMP→ smooth muscle relaxation Treatment: - PDE-5 inhibitors: effect cGMP pathway to inhibit breakdown to guanosine mono-phosphate |
|
|
Male infertility
|
- Occurs in 50% of infertile couples (30% M only, 20% M/F)
Environmental causes: smoking, drugs/alcohol, excessive exercise/testosterone, vitamin deficiency, increased scrotal temp (tight jeans), radiation, toxins, etc Hormonal causes: insufficient GnRH or LH/FSH, non-responsive to testosterone Physical: damaged ducts, varicocele, torsion, infection, retrograde ejaculation, Klinefelder’s syndrome |
|
|
Scrotal Swellings
|
Fluid filled:
- Hydrocele: serous fluid in the tunica vaginalis around the testis. Can be caused by traua, testicular torsion—peak incidence perinatal (extravaginal) and adolescent (intravaginal) - spermatocele/epididymal cyst(feels like extra testis)—connected to the epididymis, caused by an obstruction in the flow - Varicocele: dilation of veins of the spermatic cord (pampiniform plexus). Most common cause of male infertility (occur in 10-15% of men), usually left side (renal vein insertion), 10% bilateral Solid masses: - benign - malignant: tend to be within the testis parenchyma |
|
|
BPH
|
= DHT dependent growth of the prostate (in the central periurethral/transition), which increases with age due to up-regulation of androgen receptors in the medial zone
Complications: -urethral constriction resulting in obstructive symptoms: hesitancy, weak stream, straining, prolonged micturition, incomplete emptying, urinary retention - detrusor hypertrophy: reduced volume (↑frequency), bladder distension, weak points (diverticuli), vesicouretereal reflux (hydronephrosis & pyelonephritis) - Others: urinary retention/stasis, infection, stones, cancer, renal dysfunction/failure; prostate enlargement→ stretched veins→ hematuria - irritative symptoms: urgency, frequency, nocturia, urge incontinence Maintenance: reduction of symptoms/QoL, limit progression, cancer screening Tx: - meds: α-adrenergic blockers “terazosin” (dilate urethra SM), 5α-reductase inhibitors “finasteride” (reduce DHT), anti-cholinergics (reduce parasympathetic bladder contractions) - surgical: transurethral resection of the prostate (TURP), prostatectomy, bladder reconstruction (from ileum) |
|
|
Urethral obstruction
|
= obstruction of urine flow in any of 4 urethral sections: prostatic, membranous, penile/pendulous. Length in males makes it prone to obstruction
Etiologies: - Urethral stricture: almost exclusively in males, causes: trauma (straddle injury, pelvic fracture), venereal disease, prior instrumentation - Bladder cancer: mostly transitional/urothelial cell, M>F (smoking), usually present w/ painless hematuria, may cause obstruction, bleeding/clots, metastasis - Urethral cancer: F>M (↑% squamous epithelium) |
|
|
Pathologic conditions of the bladder
|
Urinary retention: obstruction, neurogenic (detrusor dysfunction), drugs
Urinary incontinence: stress (increased abdominal pressure), urge (bladder irritation→ spasm), overflow (diabetic autonomic uropathy), total (fistula, ectopic ureter drainage, congenital/traumatic) Bladder congenital anomalies - Vesicoureteral reflux: retrograde flow during micturition due to defective sphincter→ stasis, infection, chronic pyelonephritis, renal scaring - exstrophy: bladder wall opens to outside w/o skin covering - Urachal abnormalities: urine drainage out belly button, cysts, cancers - Myelodysplasia: spinal cord abnormalities (spina bifida, myelomeningioceles)→ defective sacral autonomic function - Epispadias: urethral opening along the dorsal surface of the penis, hyperspadias is ventral opening |
|
|
Conditions favoring nephrolithiasis
|
Urinary stasis
Dehydration Infection pH: low (uric acid), high (struvite, calcium phosphate) Metabolic derangements (hypecalciuria, hypocitraturia, hyperoxaluria, hyperuricosuria) Diseases/conditions: immobilization, RTA, sarcoidosis, hyperparathyroidism, short bowel, IBD (CaOx--↑Ox abs b/c reduced ileal Ca abs, Uric—diarrhea/dehydration→ low urine volume/pH) |
|
|
Urinary obstruction
|
= interruption of regular urine flow anywhere in the urinary system (unilateral or bilateral) caused by extrinsic or intrinsic factors to the GU system.
Common locations: ureteropelvic junction, ureters crossing iliac vessels, ureterovesical junction (most common) Physiologic Changes: distal tubular function affected first—impaired urine concentrating, abnormal response to ADH, decreased H+/PO4/K excretion Symptoms: acute renal colic (CVA pain + nausea from capsule stretching), edema & weight gain, abdominal distention (hydronephrosis, thin parenchyma—prone to trauma), chronic changes may be silent (↓function) Complications: urinary stasis, infection, decreased renal function, hypertension, post-obstructive diuresis w/ electrolyte abnormalities |
|
|
Diagnosis of ARF
|
Physical Exam: Assess volume status
- Orthostatic changes in BP and pulse - Skin (turgor, rash, jaundice) - Cardiovascular system: Neck (JVD), Heart (S3 gallop, friction rub) -Lungs (crackles) - G-U system: Bladder (distension), Prostate (enlargement) - Extremities (cyanosis, edema) Urinalysis: - RBCs: uniform/round (nephrolithiasis, malignancy, UTI, trauma), small/dysmorphic (glomerular disease) - WBC: neutrophils (UTI), eosinophils (AIN), - ↑renal tubular epithelial cells (ATN, AIN) - Casts: RBC (GN or vasculitis), WBC (aPN, ATN/IN), granular (glomerular/tubulointerstitial disease) - crystals: uric acid (tumor lysis), calcium oxalate (ethylene glycol), struvite (UTI), indinavir Labs: ↓BUN/Cr, ↓Uosm, ↑Una, ↑FEna, ↑FEurea Additional: foley, US, CT, intravenous pyelogram, serologies, renal biopsy Tx: renal perfusion, drug dosing, nutrition, early renal consult, cytotoxic/immune suppression, plasmapheresis, dialysis (AEIOU indications) |
|
|
Acute interstitial nephritis
|
= ARF cause by immune-mediated injury predominantly in the interstitium. Presentation typically oliguric
Causes: - drugs : NSAIDs, antibiotics, diuretics, other (allopurinol, cimetidine, phenytoin, PPIs) - Infections: bacterial pyelonephritis, viral (CMV) - Immunologic: SLE, acute rejection - Infiltrative: sarcoidosis, leukemia, lymphoma |
|
|
Acute tubular necrosis
|
= ARF caused by prolonged ischemia and/or various toxins leading to tubular damage, esp in proximal tubule or mTAL. Presentation may be either oligouric or non-oliguric ARF
- May cause decreased filtration coefficient, decreased GC pressure, increased BC pressure Pathophys (3 paradigms): Hemodynamic (vasoconstriction, tubular obstruction, urine backleak), Cell fate (injury →stunning/loss polarity), Interactive: injured cells (ischemia, toxin) →interaction →release of inflammatory mediators and cytotoxic substances Causes: - Exogenous: antibiotics, contrast, heavy metals, chemo - Endogenous: myoglobin, hemoglobin, calcium phosphate precipitation, uric acid |
|
|
Syndromes of Acute Renal failure
|
Prenrenal Azotemia = ARF caused by hypoperfusion of the glomerulus, glomerular capillary pressure is decreased
- Causes: true volume depletion (GI, renal, skin/respiratory, sequestration), decreased effective ABV (AMI/CHF, AV fistula, liver, sepsis), severe renal vasoconstriction, occlusion of renal arteries (bilateral→ anuria) Intrinsic: renal damage - Causes: acute tubular necrosis (rarely→ anuria), acute cortical necrosis (→anuria), acute interstitial nephritis, acute GN (severe→ anuria), acute renovascular disease Post renal: ARF caused by mechanical or functional obstruction to urine flow; BC pressure is increased - causes: upper (nephrolithiasis, thrombosis, fibrosis, malignancy), lower (stricture, BPH, prostate carcinoma), Neurogenic (diabetes, anticholinergics, neurologic disorders). Bilateral ureteral, bladder neck, or solitary kidney obstruction (stone, clot) → anuria |
|
|
Acute renal failure
|
= the rapid inability of the kidney over hrs/days to maintain: excretion of nitrogenous wastes (urea/creatine), fluid balance by the excretion of water, acid-base balance
Types: non-oligouric (>400 mL/d), oligouric (100-400 mL/d, Anuric <100 mL/d) Frequency: at hospital admission (1-2%), during non-critical admission (1-5%), during ICU stay 5-20% Staging: 1: Cr > 0.3mg/dL or ↑150-200%, <0.5mL/kg/hr urine for >6hrs 2: Cr ↑200-300%, <0.5mL/kg/hr urine for >12hrs 3: Cr ↑>300% from baseline or Cr 4.0mg/dL w/ acute ↑0.5mg/dL; <0.3mL/kg/hr for 24hrs or anuria for 12 hr |
|
|
Autosomal dominant polycystic kidney disease
|
Features: renal cysts (>95%), liver cysts (>80%), intracranial aneurysms, hypertension, proteinuria
Genetics: PKD1/2, mutations on primary cilia receptors, interfering with Ca/mechanomotor signaling. <5% of nephrons affect because second mutation is required for disease Dx criteria: bilateral 1cm cysts - (+) Fx: <40 3cysts, 40-60 4 cysts, >60 8 cysts - (-) Fx: 5 cysts with a consistent phenotype |
|
|
Inherited disorders of renal cell carcinoma
|
Von Hippel Lindau – Clear cell
Hereditary papillary RCC – Papillary Type 1 Hereditary leiomyomatosis RCC – Papillary Type 2 Birt-Hogg-Dube – Chromophobe (variable) Tuberous Sclerosis – Clear cell (variable) Familial Renal Oncocytoma – oncocytoma Translocation of chromosome 3 – Clear cell Medullary carcinoma – medullary Lynch syndrome (HNPCC) – transitional cell (renal pelvis) |
|
|
Birt-Hogg-Dube syndrome
|
Clinical features: fibrofolliculomas (high penetrance), pulmonary cyts, renal cell cancer & cysts, colon cancer, spontaneous pneumothorax
- Renal cancer: 15-30% incidence, Mn Dx 50.7y, multiple and bilateral w/ variable histologic appearance (even w/in families, chromophobe 34%) Genetics: BHD gene (17p11.2, highly conserved) codes for folliculin (wide tissue expression), Autosomal dominant. Management: no established guidelines, generally imaging 3-6mo for aggressive phenotypes and resection of tumors |
|
|
Hereditary Leiomyomatosis RCC
|
Clinical features: benign skin and uterine fibroids/leiomyomata, type 2 papillary renal cell carcinoma (aggressive, 10-16%, Mean Dx 44y)
Genetics: mutated FH denes for fumerase hydratase, AD is close to 100% penetrant |
|
|
Von Hippel- Lindau
|
Clinical Features : renal angiomas, CNS hemangioblastomas, endolymphatic sac tumors, pancreatic lesions (cysts, islet cell tumors, neuroendocrine tumors), renal cysts, clear cell renal
Genetics: VHL gene (3p25); mutations promote transcription of vascular growth factors and deregulation of cell cycle. Inheritance: AD (reduce penetrance, variable expression/subtypes), 20% denovo Renal cancer: clear cell, mean age of 40y, multiple and bilateral, 28-48% of patients |
|
|
Congenital anomalies of the upper urinary tract
|
Horseshoe kidney: central fusion below the inferior mesenteric artery
Retrocaval ureter: ureter deviates medially and passes behind the IVC, can cause compression and hydronephrosis Pelvic Kidney: remains in the pelvis instead of ascending Renal Agenesis: uni/bilateral failure of kidney development, associated w/ RET mutations Supernumerary kidneys: splitting of nephrogenic blastema to form extra kidneys or parts Renal Ectopia: abnormal positioning-thorax, pelvis Crossed renal extopia: one kidney is displaced to the opposite side Malrotation: failure to rotate completely, can causes drainage problems, vesicoureteral reflux, UTI Functional anomalies: dysplasia, obstruction, genetic cystic changes |
|
|
Plasma cell myeloma
|
= proliferation of B-cell clone that synthesizes and secretes a single homogenous immunoglobulin (M component) or its fragments (heavy/light chains: Bence-Jones protein in urine), “plasma cell dyscrasia”. Highest incidence age 50-60, generally poor prognosis
Pathogenesis: cytokine dependent proliferation (esp. IL-6 →poor prognosis) from neoplastic plasma and normal stromal cells in marrow. A/w a number of chromosomal abberations Presentation: - multifocal pathologic fractures/pain from destructive bone tumors (plasmacytomas) throughout the skeletal system (spine 66%, ribs 44%, skull 41%- buckshot lesions, pelvis 28%, femur 24%, clavicle 10%, scapula 10%). Also lymph and extranodal spread (skin). - Others: hypercalcemia (bone destruction), anemia/thrombocytopenia, prone to infection (despite hypergammaglobulinemia—non-functional) Histo: B cells are enlarged multinucleate, cytoplasmic inclusions (Russell Bodies). Renal: light chain cast nephropathy (eosinophilic w/ inflammatory infiltrate, occlude lumen w/ Bence-Jones proteins) Dx: serum protein electrophoresis (SPEP) to look for M component, UPEP for Bence-jones protein, bone marrow biops |
|
|
Henoch-Schonlein Purpura (HSP)
|
= non-ANCA vasculitis of unknown etiology. Primarily disease of childhood/young adults.
- Characterized by palpable cutaneous purpura, systemic manifestations (dermal, renal, joints, GI) Pathogenesis: IgA nephropathy –polymeric IgA1 deposition in arterioles leading to leukocytoclastic vasculitis and complement-mediated vascular damage (alternate pathway) Histo: IgA will deposit in dermal capillarys, in kidneys numerous glomerular dense deposits: mesangial proliferation, mesangial dense-deposit |
|
|
Microscopic Polyangitis
|
= systemic necrotizing vasculitis of small vessels, pANCA postive
- Presents as “palpable purpura” involving skin or mucous membranes, also lungs, brain, heart, GI, kidneys, nerves, and muscles. Symptoms: hemoptysis, arthralgia, abdominal pain, hematuria, proteinuria, hemorrhage, muscle pain/weakness - Can be immunologic reaction to: drugs (penicillin), microorganisms (strep), heterologous proteins, tumor antigens. Pathogenesis: triggering event causes cytokine activation of neutrophils, which adhere to vascular endothelium and degranulate releasing myeloperoxidase, and free radicals leading to DNA damage and resulting in leukocyte recruitment Histo: transmural arteritis w/ fibrinoid necrosis, leukocytoclastic vasculitis, and acute necrotizing (crescentic) GN w/ fibrinoid necrosis of the glomerulus. Pauci-immune (no IC deposit on IF/EM—neutrophil mediated) |
|
|
Wegener’s Granulomatosis
|
= granulomatous vasculitis, c-ANCA associtate, w/ classic ELK distribution, M>F average onset ~40
- Classic histo triad: acute necrotizing granulomas (upper/lower respiratory or both→ hemoptysis), necrotizing or granulomatous vasculitis (small/med vessels esp in the lungs/upper airways), acute necrotizing and/or crescentic GN. Pathogenesis: triggering event causes cytokine activation of neutrophils, which adhere to vascular endothelium and degranulate releasing proteinase 3, and free radicals leading to DNA damage and resulting in leukocyte recruitment Histo: necrotizing granulomas, transmural arteritis with fibrinoid necrosis (pink band on stain), acute necrotizing/crescentic GN w/ segmental fibrinoid necrosis of the glomerulus. Pauci-immune (no IC deposition on IF/EM—neutrophil mediated) |
|
|
SLE
|
= waxing/waning autoimmune, multi-system inflammatory disease of unknown etiology. Involves antibody formation against common cellular/nuclear components: dsDNA, RNPs, histones, cytoplasmic components, plasma proteins, surface antigens, phospholipids, etc. Characteristically affects kidneys, joints, serous membranes, blood and skin.
SOAP BRAIN: Serositis, Oral ulcers, Arthritis, Photosensitivity/Pulmonary fibrosis, Blood cells, Renal/Reynaud’s, ANA, Immunologic (anti-Sm, anti-dsDNA), Neuropsych, Malar rash Flares: may involve increase in systems involved, increase in severity of involvement, increased serologies/hypocomplementemia Diagnosis: - labs ANA (sensitive, not specific), anti-dsDNA (specific, not sensitive), anti-Sm (specific, not sensitive). For drug-induced LE, antihistone is very specific/sensitive |
|
|
Squamous & glandular metaplasia of the bladder
|
(metaplasia is benign, while de-differentiation is malignant)
Squamous: usually occurs in response to chronic inflammation - Non-keratinizing/glycogenated: more common in women (trigone area glycogenated), not a/w SCC - Keratinizing: causes hyperkeratosis, may lead to SCC. A/w smoking, schistosomiasis, arylamines (dye), phenactin, longterm catheterization Glandular: common incidental finding “cystitis cystica et glandularis,” reactive phenomona - A/w chronic cystitis, bladder exstrophy, ureteral reimplantation, neurogenic bladder. Remote risk of adenocarcinoma |
|
|
Polypoid cystitis
|
= development of grossly polypoid (edematous) or papillary lesions of the bladder in response to chronic inflammation
Causes: catheterization or fistula (cysto-rectal) Tx: remove/correct the source of the injury |
|
|
Hemorrhagic cystitis
|
= condition characterized by gross hematuria and irritive voiding (hemorrhage visible on histology)
- medical emergency, may require cystectomy Causes: cyclophosphamide, radiation, adenovirus, HSV, CMV |
|
|
Schistosomiasis
|
= inflammatory response to infection with Schistoma haematobium (blood fluke parasite)
- common in tropical areas (Africa, middle east). Worldwide is the leading cause of hematuria and bladder cancer Pathogenesis: parasite penetrates the skin, larvae enter the veins in the bladder wall (muscularis propria), larvae mature and deposit eggs which incites intense inflammatory response in the host Histologic changes: - early: necrosis, eosinophilia w/ mucosal ulceration - late: fibrosis w/ lymphocytes, histiocytes, foreign body granulomas, dystrophic calcification, squamous metaplasia → SCC |
|
|
Acute cystitis
|
= benign infection/inflammation of the bladder
- Presentation: increased frequency, lower abdominal pain, dysuria (pain/burning on urination) - Most common in young women (reproductive age) and older men (BPH→ urinary retention/stasis) - If untreated infection may ascend the ureters leading to pyelonephritis Causes: - infectious: E. coli (fecal contamination), staph saprophyticus (young women), candida or Cryptococcus (immune compromised). Indwelling catheters account for 50% of nosocomial infections - non-infectious: chemo (cyclophosphamide), radiation, trauma Tx: antibiotic (TMP-SMX), fluids |
|
|
Bladder exstrophy
|
= developmental defect in the anterior abdominal wall and the wall of the bladder in which the bladder communicates with the body surface or lies as an open sac. Due to failure of the cloacal membrane to properly differentiate.
- associated w/ glandular metaplasia (<10%)→ adenocarcinoma, and squamous metaplasia (<5%)→ SCC, due to chronic inflammation - associated with infections (UTIs) and ulceration due to exposure |
|
|
Benign vs. malignant general tumor characteristics
|
Benign:
- well circumscribed - large amount of cytoplasm relative to nucleus, no mitotic figures - cohesive, don’t exhibit metaplasia Malignant: - classic presentation triad (<10%): abdominal mass, hematuria, flank pain - invasion/metastatic potential - cellular pleomorphism - high nuclear/cytoplasm ratio, and nuclei are large and dysplastic - loss of cellular cohesion - mitotic figures |
|
|
Malakoplakia
|
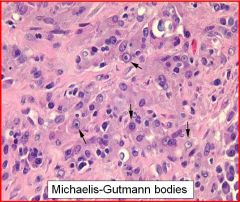
= formation of raised, yellow mucosal plaques or ulcerations anywhere in the GU tract, but most commonly the bladder.
- Caused by defects in the phagocytic or degrative function of histiocytes in response to gram-negative bacteria (E. coli, Proteus). Macrophages become filled with small, intercytoplasmic calculosperules (mineralized concretions)because of inability to degrade bacteria, forming Michaelis-Gutmann bodies - More common in immunocompromised patients (HIV, post-transplant), and women |
|
|
Urothelial carcinoma in situ
|

= neoplastic transformation of transitional cells of the urinary tract. Arises from dysplastic/atypical lesions
Characteristics: - may not full thickness, may be pagetoid (single malignant cells –look like islands in normal tissue) - CIS cells >5x stomal lymphocytes, have enlarged and hyperchromatic nuclei, and are discohesive (will begin to shed and show up in the urine) - Vessels become more prominent, prone to bleeding/hematuria - may progress to invasive urothelial carcinoma |
|
|
Urothelial Papilloma
|

= benign neoplastic lesion, typically seen in younger patients w/ no recurrence after resection, mean size 3mm
- Characterized by discrete papillary growth, a central fibrovascular core lined by urothelium of normal thickness and cytology - may progress to invasive papillary carcinoma |
|
|
Papillary Carcinoma
|

Low grade: non-invasive, rarely metastasizes, usually good prognosis
- Histo: overall orderly cellular arrangement, minimal variation in polarity, minimal atypia (scattered, enlarged, hyperchromatic nuclei), few mitotic figures High grade: invasive, aggressive, poorer prognosis - Histo: disorderly cellular arrangement (irregular clusters, fused papillae), marked atypia, numerous mitotic figures & apoptotic bodies at all levels, discohesive single cells. - Invasion to the muscularis mucosa requires cystectomy (+ prostate), usually involves blood vessel invasion, poor prognosis (<50%) |
|
|
Stage if SLE nephritis
|

- SLE nephritis is a severe complication, usually manifests <5yrs from diagnosis. Almost all SLE patients have histological involvement, 30-90% have clinical symptoms. Aggressive immunosuppression/supportive therapy have helped improve survival
Class I: minimal mesangial LN: a few IC deposits in the mesangium, little clinical evidence Class II: Mesangioproliferative LN - Histo: LM (mesangial proliferation: hypercellularity +/- increased matrix); IF (IgG and complement components mainly in the mesangium); EM (dense deposits mainly limited to the mesangium) - Clinical: little/no proteinuria +/- RBCs, normal/slight elevation creatinine, elevated ANA, normal complement, albumin, cholesterol Class III: Focal LN Class IV: Diffuse LN (>50% of glomeruli) -Histo: LM (endocapillary proliferation, extracapillary proliferation/crescents, karyorrhexis (dying nucleus fragmentation), neutrophilic exudates, fibrinoid necrosis, wire-looping, pseudothrombi), EM (subendothelial & mesangial DD’s) - Clinical: systemic symptoms (fatigue, fever, rash, arthritis, serositis, CNS, etc), abn urinalysis (leukocytes, hematuria, proteinuria, oval fat bodies, casts: granular, RBC, hyaline, fatty), labs (↑creatinine, ↑ANA/dsDNA, ↓C3/4), nephrotic syndrome Class V: Membranous LN - Histo: LM (uniform thickening of capillary loops/GBM, spikes on silver), IF (granular immunofluorescence along capillary loops), EM: subepithelial spikes/domes - Clinical: severe renal dysfunction, significant systemic activit |
|
|
Nodular prostate hyperplasia
|

= BPH involving nodule formation, can result in prostatism
- Gross: hyperplastic +/- bladder hyperplasia. Circular nodules visible in cross-section, medial lobe can push up into the bladder, peripheral zone is compressed - Histo: circular nodules consisting of hyperplastic stromal and glands, which secrete alkaline, milky fluid for ejaculation |
|
|
Prostatic carcinoma
|

= androgen dependent adenocarcinoma arising primarily in the peripheral zone of the prostate
Epi: 2nd most common cancer in men and #2 cause of cancer death Risk factors: African America (Asian is least), age >50, familial incidence, high sat. fat diet (lycopene protective?) Path: glandular formation with large nucleoli (look like eyes) Presentation: initially asymptomatic, once large can cause prostatism (via urethal compression). Can invade causing systemic symptoms Spread: direct (seminal vesicles, base of the bladder), lymphatic (obdurator lymph nodes), hematogenous (vertebrae, pelvis, proximal femur). Osseus metastases are typically osteoblastic resulting in sclerotic structures (rather than destructive) Labs: ↑OSA, ↑PAP (prostate acid phosphatase), ↑ALP (osteoblastic metastasis) Gleason grading (1-10) based on architecture rather than nuclear atypia, average presentation is 6 Tx: prostatectomy w/ radiation, anti-androgen therapy (orchiectomy, estrogen, analogs of GnRH, flutamide (androgen receptor inhibitor) |