![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
35 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Biodistribution of a drug involves |
its permeation across cellular membrane barriers. |
|
|
|
Drug permeation is dependent on |
•Solubility:Ability to diffuse through lipid bilayers (lipid solubility) is important for most drugs; however, water solubility can influence permeation through aqueous phases. •Concentration gradient: Diffusion down a concentration gradient—only free, unionized drug forms contribute to the concentration gradient. •Surface area and vascularity: Important with regard to absorption ofdrugs into the systemic circulation. The larger the surface area and thegreater the vascularity, the better is the absorption of the drug. |
|
|
|
Regarding ionisation |
••Many drugs are weak acids or weak bases, and can exist in either non ionized or ionized forms in an equilibrium, depending on the pH of the environment and the pKa (the pH at which the molecule is 50% ionized and 50% nonionized). ••Only the nonionized (uncharged) form of a drug crosses biomembranes. ••The ionized form is better renally excreted because it is water soluble. |
|
|
|
Regarding drug filtration |
••Only free, unbound drug is filtered. Both ionized and nonionized forms ofa drug are filtered. ••Only nonionized forms undergo active secretion and active or passive reabsorption. ••Ionized forms of drugs are “trapped” in the filtrate. |
|
|
|
Regarding alkalization and acidification of urine |
Acidification of urine → increases ionization of weak bases → increases renal elimination. Alkalinization of urine → increases ionization of weak acids → increases renal elimination. |
|
|
|
Absorption concerns |
The processes of entry of a drug into the systemic circulation from the site of its administration. |
|
|
|
About Intravascular administration and extravascular administration |
IA: (e.g., IV) does not involve absorption, and there is no loss of drug. Bioavailability = 100% EA: (e.g., per os [PO; oral], intramuscular[IM] subcutaneous [SC], inhalation), less than 100% of a dose may reach the systemic circulation because of variations in bioavailability. |
|
|
|
Cmax= ? |
Maximal drug level obtained with the dose. |
|
|
|
tmax = ? |
time at which Cmax occurs |
|
|
|
Lag time = ? |
time from administration to appearance in blood. |
|
|
|
Onset of activity = ?? |
time from administration to blood level reaching minimal effective concentration (MEC). |
|
|
|
Duration of action = ?? |
time plasma concentration remains greater than MEC. |
|
|
|
Time to peak = ? |
time from administration to Cmax |
|
|
|
About Bioavailability? |
It is the measure of the fraction of a dose that reaches the systemic circulation. By definition, intravascular doses have 100% bioavailability, f = 1. |
|
|
|
About Distribution |
It is the process of distribution of a drug from the systemic circulation to organs and tissue. |
|
|
|
About Distribution |
••Under normal conditions, protein-binding capacity is much larger than is drug concentration. Consequently, the free fraction is generally constant. ••Many drugs bind to plasma proteins, including albumin, with an equilibrium between bound and free molecules (recall that only unbound drugs cross biomembranes). Drug + Protein ⇌ Drug-Protein Complex (Active, free) (Inactive, bound) ••Competition between drugs for plasma protein-binding sites may increase the “free fraction,” possibly enhancing the effects of the drug displaced. Example: sulfonamides and bilirubin in a neonate |
|
|
|
About drugs with high plasma protein binding and narrow therapeutic range |
They are prone to drug interactions. (example of such drugs: warfarin, phenytoin) |
|
|
|
Special barriers to distribution include |
••Placental: most small molecular weight drugs cross the placental barrier, although fetal blood levels are usually lower than maternal(e.g., propylthiouracil [PTU] versus methimazole in pregnancy) ••Blood–brain: permeable only to lipid soluble drugs or those which are transported by facilitated diffusion or active transport (e.g., levodopa versus dopamine) |
|
|
|
About Apparent volume of distribution (Vd) |
It is a kinetic parameter of a drug which correlates dose with plasma level at zero time. |
|
|
|
Approximate Vd Values (weight 70 kg) |
Plasma volume (3 L) Blood volume (5 L) Extracellular fluid (ECF 12–14 L) Total body water (TBW 40–42 L) |
PV, BV, ECF, TBW |
|
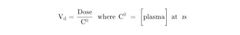
About the mathematical equation for apparent volume of distribution (Vd) |
•This relationship can be used for calculating Vd by using the dose only if one knows C0 (plasma level at zero time) ••Vd is low when a high percentage of a drug is bound to plasma proteins. ••Vd is high when a high percentage of a drug is being sequestered in tissues. This raises the possibility of displacement by other agents;examples: verapamil and quinidine can displace digoxin from tissue-binding sites. ••Vd is needed to calculate a loading dose in the clinical setting
|
|
|
|
About redistribution |
In addition to crossing the blood–brain barrier (BBB), lipid-soluble drugs redistribute into fat tissues prior to elimination. In the case of CNS drugs, the duration of action of an initial dose may depend more on the redistribution rate than on the half-life. With a second dose, the blood/fat ratio is less; therefore, the rate of redistribution is less and the second dose has a longer duration of action. |
|
|
|
General inhibitors of Cyt P450 isozymes |
antiulcer medications (cimetidine, omeprazole) antimicrobials (chloramphenicol,macrolides, ritonavir, ketoconazole) acute alcohol. |
|
|
|
About pharmacokinetics |
Pharmacokinetics is the quantitative study of drug movement in, through and out or the body The intensity of response is related to concentration of the drug at the site of action, which in turn is dependent on its pharmacokinetic properties. Pharmacokinetic considerations, therefore, determine the route(s) of administration, latency of onset, time of peak action, duration of action and frequency of administration of a drug. All pharmacokinctic processes involve transport of the drug across biological membranes. |
|
|
|
About the influence of pH |
Weakly acidic drugs, which form salts with cations, e.g. sod. phenobarbitone, sod. sul-fadiazine, pot. penicillin-V, etc. ionize more at alkaline pH and I scale change in pH causes I 0 fold change in ionization. Weakly basic drugs, which form sales with anions, e.g. atropine sulfate, ephedrine HCI. chloroquine phosphate, etc. conversely ionize more at acidic pH. Ions being lipid insoluble, do not diffuse and a pH difference across a membrane can cause differential distribution of weakly acidic and weakly basic drugs on the two sides |
|
|
|
Acidic drugs are unionized at acid gastric pH and are absorbed from stomach. True or false? |
True. Acidic drugs, e.g. aspirin (pKa 3.5) are largely unionized at acid gastric pH and are absorbed from stomach, while bases, e.g. atropine (pKa I 0) are largely ionized and are absorbed only when they reach the intestines. |
|
|
|
Ion trapping |
The unionized form of acidic drugs which crosses the surface membrane of gastric mucosal cell, reverts to the ionized form within the cell (pll 7 .0) and then only slowly passes to the extracellular fluid. This is called ion trapping, i.e. a weak electrolyte crossing a membrane to encounter a pH from which it is not able to escape easily. This may contribute to gastric mucosal cell damage caused by aspirin. |
|
|
|
T/F? Basic drugs attain higher concentration intracellularly (pH 7.0 vs 7.4 of plasma). |
True |
|
|
|
True or False? Basic drugs are excreted faster if urine is acidified |
True. Acidic drugs are ionized more in alkaline urine-do not back diffuse in the kidney tubules and are excreted faster. Accordingly, basic drugs are excreted faster if urine is acidified |
|
|
|
True or false? Transport of lipid-soluble nonelectrolytes is pH independent. |
True. Lipid-soluble nonelectrolytes (e.g. ethanol, diethyl-ether) readily cross biological membranes and their transport is pH independent. |
|
|
|
About P-glycoprotein (P-gp) |
A nonselective transporter. Encoded by the multidrug resistance 1 (MDR1) gene. P-gp is the most well known primary active transporter expressed in the intestinal mucosa, renal tubules, bile canaliculi, choroidal epithelium, astrocyte foot processes around brain capillaries (the blood-brain barrier), testicular and placental micro vessels, which pumps out many drugs/ metabolites and thus limit, their intestinal absorption, penetration into brain, testes and foetal tissues as well as promotes biliary and renal elimination. Many xenobiotics which induce or inhibit P-gp also have a similar effect on the drug metabolizing isoenzyme CYP3A4, indicating their synergistic role in detoxification of xenobiotics. |
|
|
|
Other primary active transporters of pharmacological significance |
are multidrug resistance associated protein 2 (MRP2) and breast cancer resistance protein (BCRP). |
|
|
|
About Primary active transport |
Energy is obtained directly by the hydrolysis of ATP (Fig. 2.58). The transporters belong to the superfamily of ATP binding cassettee (ABC) transporters whose intracellular loops have ATPase activity. They mediate only emux of the solute from the cytoplasm, either to extracellular fluid or into an intracellular organelli (endoplasmic reticulum, mitochondria, etc.) |
|
|
|
About Secondary active transport |
In this type of active transport effected by another set of SLC transporters, the energy to pump one solute is derived from the downhill movement of another solute. When the concentration gradients are such that both the solutes move in the same direction, it is called symport or cotransport, but when they move in opposite directions, it is termed antiport or exchange transport. Metabolic energy (from hydrolysis of ATP) is spent in maintaining high transmembrane electrochemical gradient of the second solute. The SLC transporters mediate both uptake and efflux of drugs and metabolites. |
|
|
|
About secondary active transporters |
The organic anion transporting polypeptide (OATP) and organic cation transporter (OCT), highly expressed in liver canaliculi and renal tubules, are secondary active transporters important in the metabolism and excretion of drugs and metabolites (especially glucuronides). The NaCl dependent neurotransmitter transporters for norepinephrine, serotonin and dopamine (NET, SERT and DAT) are active SLC transporters that are targets for action of drug, like tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRls),cocaine, etc. Similarly, the Vesicular monoamine transporter (VMAT-2) of adrenergic and serotoncrgic storage vesicles transports catecholamines and 5-HT into the vesicles by exchanging with H ions, and is inhibited by reserpine. The absorption of glucose in intestine, and renal tubules is through secondary active transport by sodium-glucose transporters (SGLT1 and GLT2). |
|