![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
37 Cards in this Set
- Front
- Back
|
3 General Properties of Drugs |
1. They modify functions that already exist 2. They have multiple effects almost always 3. They usually interact with something in order to exert their effect |
|
|
3 Ways Drugs modify existing functions |
1. Replace something (iron in anemia) 2. Interrupt (e.g. Ipratropium interrupts mucus production in chronic bronchitis) 3. Potentiate (e.g. cathartics before sigmoidoscopy) |
|
|
Drugs exert multiple effects: Example |
Minoxidil= lowers blood pressure + prevents hair loss |
|
|
Levels of drug activity (general to specific) |
1. Body systems 2. Component tissue 3. Cellular level 4. Molecular level * the more specific the level of understanding, the better we can use the drug and predict its SE's. |
|
|
Levels of drug activity example: Beta-Blockers |
1. Body system: reduces heart rate 2. Component tissue: negative chronotrope / prevents increase in HR in cardiac tissue 3. Cellular level: prevents elevation of cAMP (cAMP is energy source for increasing HR) 4. Molecular level: competitive antagonism of epinephrine and norepinephrine and cardiac beta 1 receptors |
|
|
2 types of receptor binding |
Agonism and antagonism |
|
|
Explain: intrinsic activity in receptor binding |
When a molecule binds to a receptor, the activity of the receptor is measured on a scale from 0 to 1. 0 = no activity (blocked) 1 = full receptor activity |
|
|
Agonism |
When a drug binds to a receptor and fully activate the receptor. Intrinsic activity = 1 |
|
|
Partial Agonism |
When a drug interacts with a receptor and produces a receptor responds that is less than 1 and more than zero. Intrinsic activity = 0 < 1 |
|
|
Antagonism |
Binding of the drug to a receptor that prevents her response to an agonist. |
|
|
Reversible v. Irreversible Antagonism |
Reversible = antagonist can be bumped off or removed by agonist Irreversible = Antagonist stays permanently bound to receptor. |
|
|
Competitive vs Noncompetitive Antagonism |
Competitive = agonist and antagonist both trying to bind the same receptor Noncompetitive = the binding of antagonist to one receptor prevents agonist from binding to and activating another receptor. Usually via changes in the membrane which subsequently blocks/distorts another receptor site. |
|
|
Pharmacological vs Effect antagonism |
Pharmacological = receptor blocking Effect = producing an opposing physiological response via a different pharmacological pathway. (e.g. cocaine + queludes) |
|
|
Ligand gated ion channels |
Transmembrane proteins that allow the exchange of ions (Na, K, Ca, Cl) in response to the binding of a chemical messenger (ligand) |
|
|
Voltage gated ion channels |
Allow the flow of ions across a membrane in response to changes in membrane potential (ion concentration) near the channel. Lots of potential receptors (i.e. "drugable targets") |
|
|
Sodium-potassium pump |
Moves both sodium and potassium across the membrane against their respective gradients. Pumping is an "active process" = requires ATP |
|
|
Enzymes |
Enzymes have binding sites that fit substrates. When bound, they effect a change in shape which subsequently creates new products from the original substrate. They can combine smaller molecules into a single molecule or break apart substrates. |
|
|
Pharmaceutics |
The science of dosage form design. How to optimally package drugs for delivery to the desired location. Helps to control SEs if precisely and accurately delivered. |
|
|
Pharmacokinetics |
Uses mathematical models to predict how the body will move, use, and modify the drug once it's in the body. i.e. tracking the absorption, bioavailability, distribution, metabolism, and excretion of the drug. |
|
|
Pharmacodynamics |
How the drug affects the body; what the drug does to the body. |
|
|
Dosage Forms: Fastest to Slowest powders coated tablets liquids, elixirs, syrups, suspensions tablet sustained release capsules enteric coated tablets |

|
|
|
Three main routes of administration |
enteral, parenteral, topical |
|
|
Enteral administration |
Anything between the lips and the anus. = oral, gastric, small intestine, rectal |
|
|
Oral administration (main points) |
Thin lining, rich blood supply
Sublingual & buccal |
|
|
Gastric (enteral) (main points) |
Large surface area, rich blood supply.
Drugs don't stay in the area long. Absorbtion is unpredictable.
Most drugs not an optimal ionic state for absorption (acidic). |
|
|
Small intestine (enteral) (main points) |
Most drug absorption occurs here.
Huge surface area, rich blood supply, basic pH. |
|
|
Rectal admin. (main points) |
Useful when do not possible (e.g. Vomiting) Similar to small intestine. |
|
|
Parenteral administration |
Used to bypass biological membrane when can't get there fast or easily enough. Includes: subcutaneous, intramuscular, intravenous, intrathecal, epidural, intraarticular. |
|
|
Topical administration (types & distribution [local v. systemic] |
Skin: local and systemic distribution Eyes: local Ears: local Nasal: local and systemic Lungs: local and systemic |
|
|
Pharmacokinetics: ADME acronym |
A = absorption D = distribution (where the drug goes, where it doesn't, and in what concentrations) M = metabolism E = Excretion (the physical removal of the drug from the body) |
|
|
Functional vs. Physical Elimination (pharmacokinetics) |
Functional elimination of a drug involves modifying the molecule such that it is deactivated or non-functional, usually in the liver.
Physical elimination refers to actually excreting the active molecule via the kidneys. |
|
|
Absorbtion: definition and main points |
Movement of drug molecules into the body. Passive vs. Facilitated diffusion. Rate of absorption can determine: -onset of action -duration of action -intensity of response * think beer bong and analogy |
|
|
Variables that affect absorption |
1. Nature of absorbing (e.g. surface surface area, intestinal epithelium vs. Skin) 2. Blood flow (sublingual vs. subcutaneous) 3. Solubility of drug (lipid vs water solubility, pH) 4. Dosage form |
|
|
Bioavailability (F) |
The percentage of the administered dose that reaches systemic circulation.
F = amount (%) of original drug that reaches systemic circulation. |
|
|
"First Pass Effect" |
Phenomenon where by the amount of drug is greatly reduced before it reaches the systemic circulation when taken PO. Typically related to being passed through the liver first.
*not the case with IV drugs (F = 100%) |
|
|
Distribution: one compartment models |

Ka= drug going in Ke= drug coming out |
|
|
Distribution: 2-compartment model |
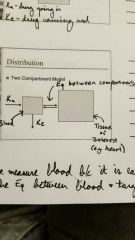
|