Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
22 Cards in this Set
- Front
- Back
|
Identify the characteristics of first-order kinetics and compare and contrast those of zero-order kinetics
|
--
|
|
|
1st order kinetics
|
-most drugs follow 1st order kinetics
-exponential -a constant fraction of drug present is eliminated per unit of time -rate of process is proportional to drug concentration Half-time (t1/2) is independent of drug concentration |
|
|
0 order kinetics
|
-Encountered only occasionally
-constant amount of the drug is eliminated per unit time, i.e., rate is independent of drug concentration Half-time (t1/2) increases with dose and is dependent on drug concentration |
|
|
Describe the cartesian and plot for first-order kinetics
|
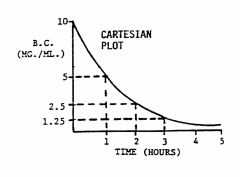
inversely parabolic
|
|
|
Describe the semi-log plot for first-order kinetics.
|
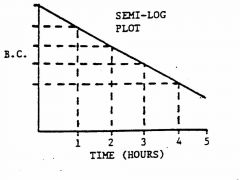
linear sloped down
|
|
|
Describe the cartesian and semi-log plots for zero-order kinetics
|
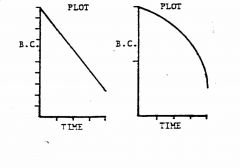
linear sloped down and parabolic
|
|
|
Describe the cartesian and semi-log plots for mass-law kinetics.
|
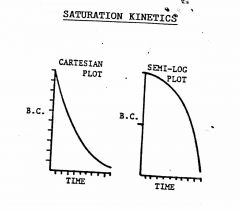
This is a mixed-order system. e.g. Renal tubular secretion of drugs for which there is a maximum tubular transport capacity. If plasma level is too high (i.e., exceeds Tm), you get zero-order until level falls, and then you get first-order kinetics.
Rate of process is a function of drug concentration, the concentration of the enzymes and their affinity |
|
|
The half-time, t 1/2, can be estimated as follows
|
t 1/2 = 0.693/Ke
• Where Ke is the elimination rate constant |
|
|
Describe the time course of plasma drug levels following IV bolus
|

Two phases:
1)Distribution phase 2) Elimination or equilibrium phase b. The exponential (first-order) processes of absorption or elimination are essentially complete after 4-5 half-times. |
|
|
Describe the time course of plasma drug levels following oral administration
|
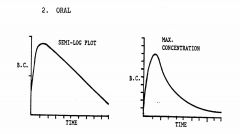
sharp increase then inversely parabolic decent (1st order) -- semilog plot shows flat slope
|
|
|
Describe the time course of plasma drug levels following IV infusion
|
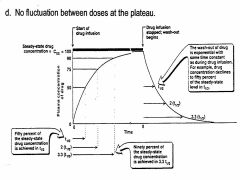
During IV infusion, the drug concentration continues to rise until the rate of elimination equals to the constant (zero order) rate of infusion, then a plateau level of drug concentration is maintained.
|
|
|
Describe the factors that affect the time course of plasma levels following IV infusion.
|
1) Plateau state:
• Attained after 4-5 half-times. • Time to plateau is independant of dose. 2) Plateau concentration: • Proportional to dose. • Proportional to half-time. • Inversely proportional to dosage interval (time in b/n dosing) |
|
|
Define bioavailability
|
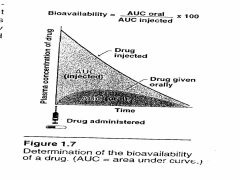
Defined as the fraction of unchanged drug reaching the systemic circulation following administration by any route. For an IV dose of a drug, the bioavailability is 100%.
Measured by comparing plasma levels (AUC) of a drug after a particular route of administration (e.g., oral) with the achieved by IV injection. AUC reflects the extent of drug absorption. |
|
|
What are the factors that influence drug bioavailability.
|
1)First-pass hepatic metabolism, e.g., propranolol
2)Solubility of drug: Drugs that are too hydrophilic or too lipophilic are poorly absorbed. For a drug to be readily absorbed, it must be largely lipophilic yet have some solubility in aqueous solution. 3)Chemical instability in gastric pH. 4)Nature of drug formulation, e.g., particle size, salt form, crystal polymorphism and the presence of inactive ingredients can influence the ease of dissolution and alter the rate of absorption. |
|
|
• Distinguish between maintenance and loading dose regimen.
|
Comparison of maintenance versus loading dose regimen:
1) Loading dose: • Immediate therapy. • May cause initial drug toxicity. 2) Maintenance dose: • Eliminates risk of toxic effects. • Permits accurate adjustment of dosage during drug accumulation. |
|
|
t 1/2 ( half life)=
|
For first-order kinetics:
*t 1/2 = 0.693/Ke Ke =rate constant of elimination |
|
|
Ke =
|
For first-order kinetics:
*Ke =0.693/t1/2 |
|
|
Vd (volume of distribution =
|
* Vd = amount of drug in body/plasma concentration
Vd is large when the drug is highly concentrated in tissues; Vd is small when the drug is extensively bound to plasma protein. |
|
|
Cl (clearance) =
|
Clearance (Cl): Total body clearance of the drug is defined as the volume of blood or plasma effectively cleared of drug by elimination(metabolism and excretion) per unit time.
It is the sum of clearance from all organs of elimination. As elimination of most drugs is carried out largely by the kidney and the liver, then * Cl = Cl renal + Cl hepatic |
|
|
IV infusion rate=
|
IV Infusion: The infusion rate equals elimination rate at STEADY STATE.
* Infusion rate= CP X Cl(amt/time)(amt/vol)(vol/time) |
|
|
intermittent injection dose =
|
*Dose = C pav X Cl X dosage interval
where C pav = average plasma concentration at the plateau |
|
|
oral maintenance and loading doses during a dosage regimen.
|
?
|