![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
396 Cards in this Set
- Front
- Back
|
Ddx of Unilateral Pediatric Nasal Mass
|
A: Encephalocele
A: Glioma A: Neurofibroma A: Nasolacrimal duct cyst A: FB/Rhinolith A: JNA A: Hemangiopericytoma A: Hemangioma A: AVM A: Polyp |
|
|
Ddx of Neonatal Nasal Obstruction |
A: Pyriform aperture stenosis
A: Midnasal stenosis A: Choanal atresia A: Nasolacrimal duct cyst A: Tumors – Encephalocele, Glioma, Dermoid, Teratoma, Hemangioma |
|
|
Percentage of laryngeal anomalies with other airway anomaly |
A: 50%
|
|
|
Nasolacrimal Duct Cyst location, pathophysiology, and treatment |
A: Location – Below inferior turbinate anteriorly
A: Pathophysiology – Proximal and distal obstruction of the nasolacrimal duct with fluid accumulation and cyst formation; 85% resolve by 9 months A: Treatment – Medical = Nasal decongestants and feeding modifications A: Treatment – Surgical = Endoscopic transnasal marsupialization, CO2 laser may be used, ophthalmology consult for intraoperative nasolacrimal duct probing stenting 3: Indications for surgery (ION) – Infection, respiratory Obstruction, Nutrition (feeding difficulties) |
|
|
Ddx of Posterior Tongue Mass & investigations |
A: Lingual thyroid
A: TGDC A: Cystic hygroma A: Valecular cyst A: Dermoid/Teratoma A: Granular cell tumor (epulis is a type on the alveolus not tongue) 3: Investigations – CT scan to define margins of the mass; TSH, T3/T4 establish thyroid function; I-123 scan or ultrasound to rule out lingual thyroid, identify other foci of functioning thyroid tissue |
|
|
Ddx Congenital Laryngeal Stridor |
A: Laryngomalacia
A: VC paralysis A: Stenosis (subglottic, tracheal) A: Subglottic Hemangioma A: Web A: Clefts A: Cysts (valecular, saccular, subglottic) A: Rings (complete tracheal, vascular) |
|
|
Ddx of Laryngotracheal stenosis |
A: Congenital –
Tracheomalacia Laryngomalacia VC paralysis Laryngeal cleft Congenital cysts Extrinsic compression, Vascular (innominate artery, right sided aortic arch, aberrant left pulmonary artery), or Mass (teratoma,lymphatic malformation, hemangioma) A: Infectious/Inflammatory – Croup Tracheitis Epiglottitis Retropharyngeal abscess GERD A: Traumatic – External compression Foreign body A: Neoplastic – Subglottic hemangioma RRP |
|
|
Seven Infectious causes of laryngeal stenosis |
A: Epiglottitis
A: Croup A: RRP A: Deep neck space infection A: Severe Laryngitis A: Bacterial Tracheitis A: Infected Laryngocele/Saccular cyst |
|
|
Discuss Junvenile Nasopharyngeal Angiofibroma (JNA) |
A: Clinical – Males, second decade (rare beyond 25 yrs), unilateral nasal obstruction and recurrent epistaxis, conductive hearing loss, dacrocystits, rhinolalia, hard and soft palate deformity, facial swelling, proptosis, cranial neuropathy, and massive hemorrhage
A: Centered at the PPF (superior border of the sphenopalatine foramen-basisphenoid) and usually extends into nasopharynx +/- the Pterygomaxillary fissure (PMF), Infratemporal fossa (ITF), Skull base, or cavernous sinus and orbit A: Path: Multiple thin walled vessels lacking smooth muscle in a fibrous connective tissue stroma with abundant mast cells, intensive immunostaining for vimentin on EM A: DO NOT BX due to the risk of bleeding and need to r/o other causes by imaging A: Tests – CT & MRI, do Angio with Embolization prior to removal; If in a female, do Karyotyping to R/O androgen insensitivity/testicular feminization A: Holman-Miller sign: the characteristic anterior bowing of the posterior maxillary wall due to the presence of a mass in the pterygomaxillary space on CT scan |
|
|
Describe the blood supply of JNA? |
A: The main supply comes from the internal maxillary artery
A: Others: ascending pharyngeal, vidian arteries, unnamed branches from the internal carotid artery (rare) |
|
|
Staging of JNA |
A: Chandler I-IV (based on NPC)
A: Sessions I-III (lateral spread to PMF and ITF) A: Radkowski I-IIIB (Sessions + extent of SB erosion) A: Fisch |
|
|
Describe the Chandler staging of JNA 1984? |
A: Stage 1 Tumor confined to nasopharynx
A: Stage 2 Tumor extends into nasal cavity or sphenoid A: Stage 3 Tumor involves the maxillary, ethmoids, infratemporal fossa, orbit, cheek, and cavernous sinus A: Stage 4 Tumor is intracranial |
|
|
Describe the JNA classification according to Sessions 1981? |
A: IA - Tumor limited to posterior nares and/or nasopharyngeal vault
A: IB - Tumor involving posterior nares and/or nasopharyngeal vault with involvement of at least 1 paranasal sinus A: IIA - Minimal lateral extension into pterygomaxillary fossa A: IIB - Full occupation of pterygomaxillary fossa with or without superior erosion of orbital bones A: IIC – ITF with or w/o cheek invasion A: III - intracranial extension |
|
|
Describe the Radkowski staging of JNA 1996? |
A: IA – Limited to nose and/or nasopharyngeal vault
A: IB – Extension to one or more sinuses A: IIA – Minimal extension to Pterygomaxillary Fossa (PMF) A: IIB – Full occupation of PMF with or without erosion of orbital bones A: IIC – Infratemporal fossa with/without cheek, or posterior to pterygoid plates A: IIIA – Erosion of skull base; minimal intracranial A: IIIB – Erosion of skull base; extensive intracranial with/without cavernous sinus |
|
|
Describe the Fisch Classification of JNA
|

A: I – Limited to nose and/or nasopharyngeal vault A: II – Extension to one or more sinuses, or the PterygomaxillaryFossa A: III – Invades the Infratemporal fossa, orbit, or parasellar areas A: IV – Extends into cavernous sinus, optic chiasm, or pituitary fossa *Classification systems of JNA |
|
|
Routes of JNA spread?
|
A: Medially: into the nasopharynx and the nasal cavity and along the vidian nerve into the floor of the sphenoid sinus A: Laterally: through the pterygomaxillary fissure leads to the infratemporal fossa A: Anteriorly: the posterior wall of the maxillary sinus is progressively pushed forward A: Superiorly (intracranial) 20-36%: From PPF through foramen rotundum From PPF → IOF → orbital cavity → SOF From ITF through foramen ovale or spinosum Through sphenoid sinus (medial to IC & cavernous sinus) Through ethmoid sinuses (anterior cranial fossa) |
|
|
Treatment strategies for JNA |
A: Hormonal therapy: flutamide (testosterone receptor blocker) or estrogen, decreases size and vascularity of tumor but due to risks and variable response not used A: XRT (30-35 Gy), generally reserved for larger and/or unressectable tumors with significant risks in a developing child A: Embolization (24-72 hours prior to excision), significantly decreased intraoperative blood loss and facilitated resection of larger tumors A: Surgery (mainstay), recurrence rates 30-50% but can spontaneously regress in some cases |
|
|
Ten possible Surgical approaches for JNA excision, from least to most invasive |
A: Endoscopic transnasal A: Transmaxillary (Transantral?) A: Transpalatal A: Lateral rhinotomy A: Medial maxillectomy A: Midfacial degloving +/– LeFort I A: Facial translocation (Maxillary swing?) A: Infratemporal fossa (Fisch C?) A: Subcranial |
|
|
Top 3 congenital laryngeal anomalies |
A: Laryngomalacia A: Vocal cord paralysis A: Subglottic stenosis |
|
|
Seven structures that can be injured during a neonatal tracheostomy |
A: Carotid (and innominate) artery A: Jugular vein A: Recurrent laryngeal nerve A: Esophagus A: Lung – Pneumothorax A: Thymus A: Larynx A: Posterior tracheal wall |
|
|
Neonatal tracheostomy safety factors intraoperatively and postoperatively |
A: Perform the tracheostomy with the neck extended using a shoulder roll A: Stay sutures in tracheal incision A: Placement of ETT before performing tracheotomy A: Keep NG tube in situ to prevent mistaking esophagus for trachea A: Postoperative observation in PICU A: Tracheostomy set at the bedside A: Flexion of neck while applying ties A: Do not tack skin edges together to avoid subcutaneous emphysema A: CXR in recovery room to verify tube position and to R/O pneumothorax A: Always keep at bedside – Trach set with Hemostat, Suction, Same size and smaller trach tubes A: First tube change at 5-7 days |
|
|
Grading Firm Mature SGS, Myer & Cotton (1994) |
A: Using cuffless pediatric ET tube A: Assess air leak. If < 10cm H20, upsize tube; if btwn 10-25 cmH20 (compare to expected ETT); if > 25 cmH20, downsize tube A: Comparing to expected size ET tube for patient age deduction % of lumen obstruction from the above 3: Usefulness in prognosis for decannulation, and number of operations required to decannulation |
|
|
Cotton-Myer grading of SGS |
A: I) 1-50% A: II) 51-70% A: III) 71-99% A: IV) 100% |
|
|
McCaffrey system classifies laryngotracheal stenosis |
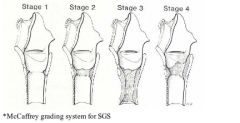
A: Stage I – confined to the subglottis or trachea, <1 cm long A: Stage II – isolated to the subglottis, >1 cm long A: Stage III – subglottic/tracheal lesions not involving the glottis A: Stage IV – lesions involve the glottis |
|
|
Bogdasarian classification of adult posterior glottic stenosis |
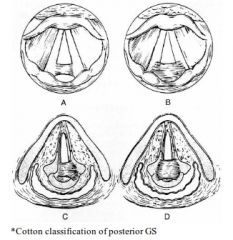
A: Type 1: Interarytenoid adhesion (with posterior sinus tract in Cotton classification)
A: Type 2: Posterior commisure stenosis A: Type 3: Posterior commissure stenosis with unilateral cricoarytenoid ankylosis A: Type 4: Posterior commissure stenosis with bilateral cricoarytenoid Ankylosis |
|
|
Etiology/predisposing factors for acquired SGS in adults (10)
|
A: Intubation-related (>90%, 1-8% incidence): oversized, repeated, shearing motion (agitation), route, and duration A: Iatrogenic trauma (laser surgery, high tracheotomy, cricothyrotomy) A: External laryngeal trauma A: Burn (inhalational/thermal/chemical/radiation) A: Gastroesophageal reflux A: Infection (Primary or Superimposed bacterial or fungal infection) A: Neoplasms (Benign or Malignant, Intrinsic or Extrinsic) A: Autoimmune (Wegeners, Sarcoidosis, SLE) A: Inflammatory disease (Sarcoidosis, Relapsing Polychondritis) A: Idiopathic SGS |
|
|
Four Preventative measures for avoiding SGS |
A: Smaller ETT without compromising safe ventilation (air leak at <25 cmH2O) A: Diagnosing (pH probe) & treating LPR A: Prophylactic Antibiotics when tracheotomy is performed following prolonged/traumatic intubation A: Prolonged intubation up to 6 months preferred over tracheostomy in neonates |
|
|
Histopathologic classification of Congenital SGS |
A: Soft tissue – Granulation tissue, submucosal gland hyperplasia, submucosal fibrosis A: Cartilaginous – Normal shape (cricoid small for infant’s size) A: Cartilaginous – Abnormal shape (elliptical shape, large anterior lamina, large posterior lamina, generalized thickening, submucus/incomplete laryngeal cleft, other) A: Cartilaginous – Trapped first tracheal ring A: Combined stenosis |
|
|
Definition of Congenital SGS (vs Acquired SGS) |
A: No history of ETT or laryngeal trauma |
|
|
Which has worse symptoms and prognosis: Congenital or Acquired SGS? |
A: Acquired 3: Congenital tends to improve with growth of the child |
|
|
Normal term subglottis |
A: 4.5 – 5 mm (4 mm in premature; BB says 3) A: Size 3 ETT |
|
|
SGS at term & premie? |
A: <4 mm & <3.5 mm (BB says <3mm) |
|
|
Rule for choosing the appropriate ETT size |
A: Age/4 + 4 or (Age+16)/4 |
|
|
By what percentage will 1 mm of subglottic edema reduce the airway in a neonate? |
A: 67% (BB says ~60%) |
|
|
Pediatric bronchoscope sizes (outer diameter) |
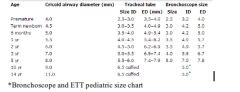
A: Premie - 2.5 (3.7) A: 0-3 months (Term)- 3.0 (5.0) A: 3-18 months - 3.5 (5.7) A: 1-3 years - 3.7 (6.3) A: 2-6 years - 4.0 (6.7) A: 5-10 years - 5.0 (7.8) A: 10-16 years - 6.0 (8.2) 3: Outer diameter (OD)= inner diameter (ID) + 0.8 3: ID is the same as the size of an ETT while they’re two different values in bronchoscopes (see below) |
|
|
Pediatric esophagoscope/laryngoscope sizes |
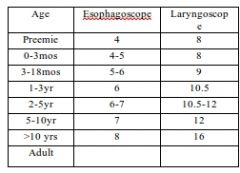
|
|
|
Smallest bronchoscope able to accommodate peanut grasper
|
A: 3.5 |
|
|
2 alternatives to ETT in the airway management of known SGS cases |
A: Laryngeal mask airway A: Heliox A: TIVA |
|
|
Management options for SGS |
A: Observation – Grade I-II with minimal symptoms & reliable follow up, especially congenital, repeat bronch q3-6 months A: Medical – Anti-reflux A: Tracheotomy A: Endoscopic procedures – Balloon dilatation, Laser, mitomycin C A: Open reconstructive procedures – Expansion procedure (LTP/LTR, single stage or with trach and Stent: Anterior w/wo Posterior w/wo Lateral cricoid split, anterior and/or posterior cartilage Graft) Segmental resection (Cricotracheal resection & anastomosis, primary, salvage, extended with expansion, arytenoid lateralization or arytenoidectomy, stents) |
|
|
Contraindications to airway surgery |
A: Absolute – A: Tracheotomy dependent (aspiration, severe BPD) A: Severe GER refractory to surgical and medical therapy A: Unfit for GA A: Relative – A: Steroid use A: Diabetes A: Cardiac, renal or pulmonary disease |
|
|
Five Indications of Laser for SGS |
A: Early stenosis A: Grade I, II A: Granulation tissue A: Thin webs A: Crescent-shaped bands 3: “Early mild soft thin crescents” |
|
|
Eight Contra-indications of endoscopic laser for SGS |
A: Circumferential thick (cicatricial) scarring A: Length >1 cm A: Laryngotracheal stenosis A: Posterior glottic stenosis with arytenoid fixation A: Previous failure A: Previous severe bacterial infection associated with tracheostomy A: Exposure of cartilage during CO2 laser excision predisposing to chondritis A: Loss of cartilaginous framework |
|
|
Indications & Contraindications for Anterior Cricoid Split |
A: Indications – Failure of extubation 2 times in neonate/young child, congenital small cricoid in older child A: Contraindications – Short duration of extubation before reintubation (hours), Peak airway pressure > 35 mm Hg |
|
|
Seven selection Criteria for Anterior Cricoid Split |
A: Weight > 1500 gm A: Failed extubation twice 2ndry to laryngeal pathology A: No acute respiratory tract infection A: O2 requirement < 30% A: No ventilation support for at least 10 days A: No antihypertensive medications at least 10 days A: No CHF for at least 1 month 3 broad catagories: Airway, ventilatory, cardiac H20 UVWX Heart failure, Hypertention, O2, URTI, Vent support, Weight, Extubation |
|
|
How much distraction of the cricoid is required for a cartilage graft to be placed in the anterior split |
A: 3mm |
|
|
Most common techniques for laryngotracheal reconstruction (LTR) |
A: Anterior cartilage graft + tracheotomy + no stent A: Short term stenting (4-6 weeks) + cartilage grafts (anterior &/or posterior) A: Long-term stenting (several months) +/– cartilage graft A: Single stage LTR (SSLTR) – cartilage grafts + brief period of nasotracheal intubation (7 - 10 days for ant graft, 10 - 14 days for post graft, older = shorter) |
|
|
Four indications for 2-Step LTR |
A: Severe stenosis A: History of reactive airway A: Poor pulmonary function A: Inadequate intensive care facilities |
|
|
Three indications for LTR with Division of the Posterior Cricoid lamina |
A: Posterior Glottic/Subglottic stenosis A: Complete Glottic/Subglottic stenosis A: Significant Cricoid deformity |
|
|
Four indications for Cartilage Grafting in the posterior glottis and subglottis |
A: Posterior Glottic/Subglottic stenosis A: Isolated Subglottic shelves A: Circumferential Subglottic stenosis A: Total or near total obstruction at the glottic or subglottic level |
|
|
Five indications for Long-term Stenting in pediatric airway reconstruction |
A: Posterior cricoid split without cartilage grafting A: Lack of airway wall Rigidity A: Keloid formation A: Severely altered anatomy by stenosis or surgery A: Unstable cartilage grafts 3: “Posterior Rigid Keels are Severely Unstable” |
|
|
Five types of Stenting |
A: Aboulker or Cotton-Lorenz stent (rigid Teflon – polytef II, hollow lumen) A: Montgomery T tube (hollow silicone) A: Montgomery laryngeal stent (solid silicone) A: Single stage LTP (ETT used as alternative to stenting) A: Finger cot A: Silastic sheet / Swiss roll |
|
|
Four advantages of Cricotracheal Resections & Thyrotracheal Anastomosis |
A: Safe effective treatment for Severe SGS A: Results are Superior to similar cases done by LTR techniques A: Voice quality results are better (preserves voice) A: No interference with normal growth of Larynx 3: Contraindication of CTR: subglottic scarring within 3 mm of vocal cords |
|
|
Two disadvantages of Cricotracheal Resections & Thyrotracheal Anastomosis |
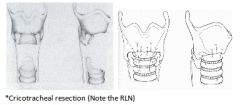
A: Possibility of injury to the Recurrent Laryngeal Nerve (lateral cricoid dissection is performed in subperichondrial plane & lateral resection is anterior to the Cricothyroid joint) A: Possible partial Dehiscence at anastomotic site resulting in Restenosis (laryngeal release only if >5 tracheal rings resected) |
|
|
Three options for Post-operative airway support for Glottic Edema following Cricotracheal Resections & Thyrotracheal Anastomosis
|
A: Short term ET intubation A: Montgomery T tube stenting for older child (4-6 weeks) A: Distal tracheotomy (4-6 weeks) |
|
|
Two indications for Four Quadrant Cricoid Split |
A: Grade III & IV A: Congenital elliptical cricoid 3: Procedure – Division of ant. + post. walls (+/- grafts) + lateral walls of cricoid anterior to inf. cornu of thyroid & extraperichondrial externally to avoid RLN, Aboulker or Cotton- Lorenz Stent for 6 months (very unstable airway) |
|
|
Idiopathic subglottic stenosis (ISS) |
A: Rare inflammatory process of unknown cause A: Limited to subglottis & upper 2 tracheal rings A: Young female >85% (estrogen altering wound healing response?) A: Surgery is the main treatment modality (endoscopic laser with mitomycin-c for < 1cm, open laryngotracheal surgery (CTR) for thicker complex scar) |
|
|
Safe length of time for intubation in adults, children, and neonates |
A: Adults – 5-10 days A: Children – up to 50 days A: Neonates – up to 6 months |
|
|
Ddx of Pediatric Lateral neck mass (6) |
A: Branchial anomaly A: Laryngocele A: Pseudotumor of infancy A: Hemagioma A: Lymphatic malformation A: Thymic cyst |
|
|
Ddx of Pediatric Midline neck mass |
A: TGDC A: Dermoid A: Teratoma A: Plunging ranula A: Thymic cyst A: Hemagioma A: Lymphatic malformation |
|
|
Histology of Thyroglossal duct epithelium |
A: Squamous A: Respiratory A: Thyroid follicles and colloid |
|
|
What are the most common H&N peds malignancies in general and rank by age? |

|
|
|
Lymphoma pearls
|
A: 60% of pediatric lymphomas are NHL but Hodgkin’s is more common in the H&N (see above) A: Presentation: NHL in the H&N is seen in 5-10% of children and most often extranodal involving Waldeyer’s ring, salivary glands, larynx, sinuses, orbit and scalp that rapidly progresses but can lead to asymptomatic cervical LN HD will present with asymmetric lymph node enlargement above the diaphragm in about 90% of cases A: EBV is associated with Burkitt’s (90% of endemic BL & 20% of sporadic BL) Hodgkin’s lymphoma in 19-59% A: Risk factors for NHL: AIDS Immunosuppressive therapy Chemotherapy Congenital immunodeficiency syndromes (Wiskott-Aldrich syndrome, ataxia-telangiectasia, X-linked lymphoproflerative disorders) |
|
|
Pathognomic cell in Hodgkin’s lymphoma |
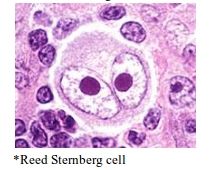
A: Reed Sternberg cell |
|
|
Rye classification of Hodgkin’s lymphoma
|
A: Nodular sclerosis (60%) – nodules of lymphoid infiltrates, lacunar variants of RS cells A: Mixed cellularity (30%) – pleomorphic lymphocytes, more numerous RS cells A: Lymphocyte depleted (6%) – paucity of lymphocytes, diffuse fibrosis and bizarre RS cells; worse prognosis A: Lymphocyte predominant (3%) – rare Reed-Sternberg cells, favorable prognosis |
|
|
Ann Arbor staging of Hodgkin’s lymphoma |
A: I – Single LN region or extralymphatic site (IE) A: II – 2+ LN regions or EL sites (IIE) on same side of diaphragm A: III – Nodal regions, EL sites (IIIE), or spleen involvement (IIIS) on both sides of diaphragm A: IV – Disseminated disease A: A (absence) or B (presence) of unexplained weight loss >10% of total body weight, unexplained fever >38, night sweats |
|
|
Treatment of Hodgkin’s lymphoma |
A: Early disease (I & IIA) – XRT, 10 year survival 90%, 10 year relapse free survival 75-80%, chemo given for mediastinal disease A: Advanced disease – Combination chemotherapy +/- XRT: MOPP (nitrogen mustard, vincristine, procarbazine and prednisone) ABVD (Adriamycin [doxorubicin], bleomycin, vinblastine, dacarbazine) Stanford V (doxorubicin, vinblastine, mustard, bleomycin, vincristine, etoposide, prednisone) Complete response rate 44-87%, long term disease free survival rate 50% A: Children – Combined modality therapy equally effective while causing less growth impairment |
|
|
Working classification of non-Hodgkin’s |
A: Low grade – Small lymphocytic, Follicular small cleaved, Follicular mixed. (SMALL) A: Intermediate grade – Follicular large, Diffuse small cleaved, Diffuse mixed, Diffuse large cell (most common in H&N). (DIFFUSE) A: High grade – Small noncleaved (Burkitt), Immunoblastic (Large cell), Lymphoblastic. (BURKETTS OR LARGE) |
|
|
Staging of non-Hodgkin’s disease |
A: I – Single LN region or ES site with exclusion of mediastinum and abdomen A: II – Single ES site with regional node involvement; 2+ LN regions or ES sites on same side of diaphragm A: III – Nodal regions or ES sites on both sides of diaphragm; any intrathoracic, paraspinal, epidural tumor A: IV – Disseminated disease or any of above with initial involvement of CNS, bone marrow, or both |
|
|
Describe the high grade NHL lesions |
A: >90% of children have high grade lesions A: Burkitt lymphoma (small noncleaved) – Diffuse B-cell malignancy, classic starry sky pattern of phagocytic histiocytes and tumor cells, Genetics: translocation of myc gene from chromosome 8 to 14 in 80% A: Lymphoblastic – Immature T-cell origin, small lymphoblasts with round/convoluted nuclei, distinct nuclear membranes, basophilic cytoplasm A: Immunoblastic/Large cell – Heterogeneous group of lymphocytic & histiocytic tumors; 80% of adults are B-cell in origin; in children equal numbers originate from T-cell, B-cell or indeterminate origin |
|
|
Discuss the clinical assessment for non-Hodgkin’s lymphoma after LN biopsy |
A: H&P with direct laryngoscopy A: CBC/Diff, LFT’s, LDH A: CT neck, chest, abdomen & pelvis A: Barium swallow (3- 11% of patients with Waldeyer ring lymphoma will have an associated GI lesion) A: Bone marrow biopsy (18% of patients with extranodal H&N lymphoma involved) A: Lumbar puncture (at risk of CNS involvement: High grade lymphoma, Intermediate grade lymphoma of the paranasal sinuses, bone marrow, testes, paraspinal areas) |
|
|
Discuss treatment of NHL? |
A: Combination chemo: CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) A: For extensive disease: methotrexate, ifosfamide and other chemo agents maybe added +/- XRT +/- Surgery for airway issues |
|
|
Most common primary sites in the head and neck for |
Rhabdomyosarcoma in descending order (ONES)? A: Orbit (25-30%) A: Nasopharynx A: middle Ear/mastoid A: Sinonasal cavity 3: 35-40% of all cases occur in the head and neck |
|
|
Most common metastatic sites for head and neck Rhabdomyosarcoma |
A: Lungs A: Bone A: Bone marrow |
|
|
Classification of rhabdomyosarcoma according to site in the head and neck? |
A: Orbital, most common, treated with CRTx (No Sx) A: Parameningeal sites (nasopharynx/nasal cavity, the middle ear, the paranasal sinuses, and the infratemporal fossa/pterygopalatine space), worse prognosis due to skullbase/IC involvement in 65-80%, Treated with CRTx +/- Sx A: Non-parameningeal sites: Superficial sites: scalp, cheek, and external ear, or Deep structures: parotid gland, larynx, oral cavity, and oropharynx. More amenable to surgical excision 3: Orbit and non-PM are favorable sites while PM sites are unfavorable |
|
|
Histologic classification of Rhabdomyosarcoma |
A: Embryonal (60-70%) Most common in infants and children in the H&N Intermediate prognosis (Best prognosis in spindle cell and botryoid subtypes) Histo: Small round to spindle shaped cells (strap cells) with eosinophilic cytoplasm (blue) Botryoid subtype appears as a cluster of grapes & arise under the mucosal surface of body orifices such as the vagina, bladder, nasopharynx Spindle cell subtype usually involves paratesticular site A: Alveolar (20%) Most common in adolescents in the extremities & trunk Poor prognosis Histo: clusters of small round cells with fibrous septae ( resembles lung alveoli) in > 50% of the tumor (if <50% → embryonal) A: Pleomorphic/Anaplastic (5%) Almost exclusive in adults in the extremities & trunk Poor prognosis Histo: Greater degree of nuclear atypia, multinucleation and pleomorphism. Some consider this a subtype of embryonal RS in peds and a form of malignant fibrous histiocytoma in adults and deleted it as a type on its own in the new classifications A: A fourth class may be Undifferentiated, poor prognosis |
|
|
Immunohistochemical stains for rhabdomyosarcoma? |
A: myogenin, muscle specific actin, and desmin |
|
|
How to stage rhabmyosarcoma? |
A: Pre-operative staging, using the TNM classification, is commonly referred to as “stage” A: Post-operative staging system is commonly referred to as “group” depending on amount of residual disease post Sx A: Then assign a Risk Group: Determined by Stage, Group, and histology |
|
|
TNM Staging of rhabdomyosarcoma from IRS-IV |
A: T staging T1 – Confined to anatomic site of origin T2 – Extension and/or fixation to surrounding tissue A ≤5cm in diameter B >5cm in diameter A: N staging N0 – Not clinically involved N1 – Clinically involved Nx – Clinical status unknown A: M staging M0 – No distant metastases M1 – Distant metastasis A: Stages I – Head and Neck (Orbit, non-PM); T1 or T2, A or B; any N; M0 II – Parameningeal; T1 or T2, A; N0 or Nx; M0 III – Parameningeal; T1 or T2, B; any N; M0 IV – All sites; T1 or T2, A or B; any N; M1 3: Groups and risk classification can be seen in staging pdf |
|
|
Discuss general treatment of Rhabdomyosarcoma |
A: Surgical excision Should be done if feasible with 2 cm margin Not recommended if produces significant morbidity (functional or cosmetic) or no increase in survival seen post excision compared to CRTx (e.g orbit) Can be used for salvage A: Induction chemo in all cases: vincristine and dactinomycin +/- cyclophosphamide (VAC) A: Radiation therapy (40-50Gy) is used in cases with gross or microscopic disease after surgery or in cases in which surgery is not feasible A: Neck should NOT be treated (Sx or RT) unless N+ |
|
|
Discuss Post-transplantation lymphoproliferative disorder (PTLD)? |
A: Umbrella term for all abnormal proliferations of lymphoid tissue in the transplant recipient, ranging from lymphoid hyperplasia to NHL A: PTLD usually manifests with B-cell proliferation induced by EBV; this proliferation is left unopposed by the pharmacologically suppressed T-cell system A: Risk factors: degree & type of immunosuppression, EBV seronegative status at time of transplant, young age, donor-recipient mismatch & GVHD, T-cell depletion or use of anti T-cell monoclonal antibodies A: Tonsillar hypertrophy, adenoid enlargement +/- cervical adenopathy is a common presentation in the H&N A: Management options: Biopsy of tissue to diagnose & exclude lymphoma Excision of obstructing lymphoid tissue (T&A), can be curative Reduction or cessation of immunosuppression Antiviral treatment with acyclovir or ganciclovir to control Epstein-Barr virus (EBV) replication Interferon alpha IV immunoglobulin Immunotherapy with donor t-lymphocyte infusion Chemotherapy +/- RT for lymphoma cases Surgery to remove the transplanted organ |
|
|
Discuss Atypical mycobacterial infection |
A: Acid fast gram positive obligate aerobes that can be found in the environment in soil, water, vegetables, and even in domestic animals and dairy products A: Most common are M. Avium intracellulare, M. kansasii, M. scrofulaceum A: Childhood disease between ages 1-5 years A: Submandibular region > pre-auricular > parotid region A: Nontender, slowly enlarging, skin fixation common with a violaceous hue A: Corneal ulceration is most common H&N manifestation A: Few systemic effects, rare pulmonary involvement A: PPD (5 units) intradermally → negative or weekly positive (If strongly positive may suggest typical TB → CXR) A: Ziehl-Neelson stain shows AFB & Lowenstein-Jensen medium for c/s (2-8 weeks incubation period) A: Antibiotics: Less cure rate compared to Sx, adjunct to Sx, if severe adenopathy, residual/recurrent disease, immunocompromised pt , disseminated disease mono, dual or triple therapy: Clarithromycin or azithromycin +/- Ethambutol +/- Rifampin (or rifabutin) for Disseminated disease Used for 6-12 months A: Incision and drainage may cause fistulization A: Curettage or excision with skin is the main modality of treatment (Caution in regards to marginal mandibular nerve) |
|
|
Infantile Hemangioma age of appearance, Gender predilection, percent Multiple, Cellular findings, site in the airway and syndrome associated |
A: First 6 weeks A: Female: Male = 3-6:1 A: Percentages: 20% Multiple 50% with a Subglottic hemangioma will also have a cutaneous lesion, but the converse in only true in 1-10% However, 63% of young children with hemangiomas in four or more sites in the beard distribution have airway lesions A: Proliferating Endothelial cells and increased Mast cells, stains positively for glucose transporter 1 (GLUT-1) A: Predilection for the Left Posterolateral subglottis A: PHACES syndrome (posterior fossa malformations (P), segmental facial hemangiomas (H), arterial anomalies (A), cardiac defects (C), eye abnormalities (E), and sternal defects (S)) 3: usual size 0.5-5 cm (up to 20 cm) |
|
|
Stages of Infantile Hemangioma evolution |
A: Proliferative – 6-12 months A: Involuting – 50% by 5 years, 70% by 7 years A: Involuted – Redundance, scarring, telangiectasias 3: Specific Markers of proliferation: Serum and urinary vascular endothelial growth factor (VEGF), Urinary beta-fibroblast growth factor (b-FGF), Urinary matrix metalloproteinases (MMPs) |
|
|
Indications for treatment of Hemangioma (VASCO)? |
A: Impaired Vision or Hearing A: Airway compromise A: Impaired Swallowing A: Cosmesis (massive, ulcerating, disfiguring) A: High Output Cardiac failure 3: Complications of Hemangiomas are the above + Kasabach-Merritt Sx (hemangioma-thrombocytopenia) |
|
|
Treatment options for Hemangioma |
A: Observation A: Standard of care = Tracheostomy and wait for involution A: Oral steroids (2-3 mg/Kg/d PO x 7 days then R/A, if responsive, taper over 4-6 weeks up to 10 months) A: Intralesional steroids (Triamcinolone 40 mg or Betamethasone 6 mg q4-6 weeks x 1-5, avoid periorbital) A: IFN-Alpha 2a or IFN-Alpha 2b (unresponsive to steroids, can be daily S/Q x 6+ months) A: Surgery (endo or open cold knife with cricoid split, Microdebrider, CO2 /KTP /Nd:YAG / Pulse dye laser) A: Photocoagulation (q4-6weeks, early proliferative phase, superficial lesions) A: Cryotherapy A: Tracheostomy or Laryngotracheoplasty A: Propranolol: Mode of action: BetaBlocker, vasoconstriction, down-regulation of angiogenetic factors such as VEGF and bFGF, up-regulation of apoptosis of capillary endothelial cells, inhibiting the expression of MMP-9 & HBMEC Side effects: bradycardia, hypotension, hypoglycemia, rash, gastrointestinal discomfort/reflux, fatigue and bronchospasm CI: Large hemangiomas are at risk for high-output cardiac compromise if used, renal or hepatic dysfunction, underlying cardiovascular disease, asthma, diabetes, glaucoma or allergy Dose: initiated with dose of 0.5-1 mg/kg/day (divided PO TID) with cardiac & BS monitoring x 48 hrs, If tolerated → increased to 2-3 mg/kg/day x 6-12 months then tapered off |
|
|
Subglottic Hemangioma – which type is safe to biopsy and use CO2 laser on |
A: Capillary – Less colour to lesion, smaller vessel size, therefore able to use laser A: Cavernous – Dark red/blue, bleed with biopsy, and difficult to control with CO2 laser |
|
|
Complication of laser of subglottic hemangioma |
A: Subglottic stenosis |
|
|
Other vascular neoplasms? |
A: Rapidly involuting congential hemangioma (RICH): - Present at birth and involute in the first yr, GLUT1 –ve - Tx: observation A: Non-involuting congential hemangioma (NICH): - Present at birth and persist, GLUT-1 –ve - Tx: Laser and surgical Tx A: Lobular capillary hemangioma (pyogenic granuloma) A: Kaposiform hemangioedothelioma A: Tufted angioma A: Angiosarcoma |
|
|
Kasabach-Merritt phenomenon |
A: Kaposiform hemangioendothelioma or tufted angioma A: Sequestration of platelets, ecchymoses A: Heparin contraindicated A: Tx: Supportive care, steroids, IFN-A2a, PRBCs for anemia, limit blood products unless bleeding, RT or embolization maybe used |
|
|
Classification of vascular malformations |
A: Low flow (capillary, venous, lymphatic, combinations) A: High flow (arterial, arteriovenous) |
|
|
Discuss vascular malformations? |
Capillary malformations Dilated capillaries (Port-wine stain) If involving upper face & eyelid → R/O Sturge- Weber Sx (MRI brain & ophtho consult); glaucoma, sz, mental retardation Lesions progressively darken and thicken Serial laser therapy the management of choice (argon, pulsed tunable dye) Surgical excision possible; watch for hypertrophy or unpredictable pigmentation Venous malformations Abnormally tangled vessels with slow flow Compressible, enlarge with valsalva or gravity CT may show calcified phleboliths May lead to consumptive coagulopathy Tx: Symptom management and surgical excision/ laser / or sclerotherapy in selected lesions Lymphatic malformations Divided into Macrocystic (>2cm3) Usually infrahyoid and lateral to canthus Resolve spontoneusly if not septated + posterior neck + InfraH Resolves with sclerotherapy or Sx otherwise Microcystic (<2cm3) or Mixed Usually suprahyoid and midface Involves oral & OP mucosa (infiltrative) Often associated with complications: A/W, speech, feeding, bleeding Responds poorly to Tx Associated issues: Sudden ↑ in size occur w/ trauma, hemorrhage or infx of cystic spaces → broad spectrum Abx Lymphopenia can occur Bony overgrowth or resorption (Gorham Syndrome) Associated Syndromes: Turner syndrome, Down syndrome, Klinefelter syndrome, and trisomy 18, 13, Noonan syndrome, Fryns syndrome, multiple pterygium syndrome, and achondroplasia MRI findings: septated masses w/ low intensity on T1 high intensity on T2 w/out flow voids If detected on prenatal U/S with possible A/W issues → EXIT procedure Surgical resection mainstay of therapy +/- trach Cold knife laser (CO2, Nd:YAG) if oral cavity & tongue Considered if minimal morbidity and unsuccessful other treatments No consensus on timing Sclerothrapy (e.g. OK-432): Ineffective for microcystic disease & good response rates for macrocystic disease Aspirate macrocyst then inject agent under fluro Other treatment: RT, RF ablation, Sildenafil (new trials) AV malformations Abnormal communications b/w arteries & veins, bypass capillary bed, high flow Intracranial > extracranial Characteristically a pulsatile mass w/ an assoc thrill or bruit on auscultation or Dopler, warm, on cheek or auricle Stages: dormancy, expansion, destruction & CHF Dx with MR or CT angio MRI: no enhancement on T2 flow voids present on both T1 & T2 Cx: skin necrosis ulceration bleeding heart failure Tx: Nothing required for asymptomatic lesions symptomatic lesions require embolization alone if in bone or preoperative embolization and surgical resection of nidus if involving soft tissue |
|
|
De Serres Lymphatic malformation staging? |
A: Stage I - Unilateral infrahyoid (17% Complication rate) A: Stage II - Unilateral suprahyoid (41% Cx rate) A: Stage III - Unilateral infrahyoid and suprahyoid (67% Cx rate) A: Stage IV - Bilateral suprahyoid (80% Cx rate) A: Stage V - Bilateral infrahyoid and suprahyoid (100% Cx rate) 3: Modified Seattle classification added: Stage VI Bilateral infrahyoid, Stage VII Retropharyngeal, M for mediastinal extension |
|
|
Options for Sclerotherapy |
A: Effective for lymphatic and venous malformations A: Ethibloc (95% ethanol with starch) A: Sodium tetradecyl sulphate A: OK-432 (lyophilized low-virulence Strep Pyogenes, macrocystic = 92% response microcystic = 44%) A: Bleomycin A: Doxycycline |
|
|
MRI Characteristics of vascular anomalies?
|
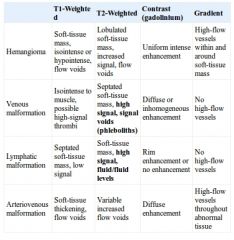
|
|
|
Differences between Hemangioma & vascular malformations?
|
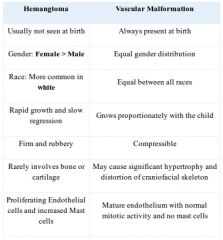
|
|
|
9 anatomic relationships of a 2nd branchial arch anomaly
|
A: External opening along lower third of SCM A: Internal opening associated with posterior pillar in tonsillar fossa A: Deep to platysma, CN VII, external carotid A: Superficial to stylopharyngeus, CN IX, X, XII, & internal carotid |
|
|
Rule of branchial arch anomaly relationships |
A: Run deep to own arch structures A: Run superficial to next arch structures |
|
|
Which cranial bones are formed by Endochondral ossification (i.e. the others are all intramembranous) |
A: Hyoid bone A: Inferior turbinate A: Styloid process A: Petrous Temporal A: Occipital A: Ethmoid A: Mastoid A: Sphenoid A: “HIS POEMS” |
|
|
Triad seen in Pierre-Robin |
A: Micrognathia A: Cleft palate A: Glossoptosis 3: If isolated (non-syndromic) mandible catch up growth happens in first year and attains normal profile in 5-6 yrs. If syndromic, this persists |
|
|
Percent of Robin sequence associated with a syndrome |
A: 50-80% A: Stickler A: VCFS 22q11 A: Others: Treacher Collins, trisomy 11q syndrome, trisomy 18 syndrome, Möbius syndrome, and CHARGE association |
|
|
Management options for respiratory distress in Pierre-Robin patient |
A: Medical – Prone position, McGovern nipple, Nasopharyngeal airway, NG tube, Intubation (difficult), NIPPV A: Surgical – Tracheostomy, Subperiosteal Floor of mouth release, Glossopexy, Tongue-lip adhesion (Routledge), Distraction osteogenesis, CP repair |
|
|
Features of Stickler syndrome (hereditary progressive arthro-ophthalmopathy)? |
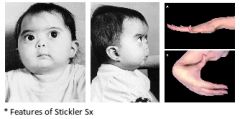
A: AD mutation of COL2A1 gene, chromosome 12, for type II collagen A: Robin sequence, mid face hypoplasia A: Eye – Myopia, cataracts, & retinal detachment A: Joint – Hypermobility & enlarged joints, early arthritis, occ. spondyloepiphyseal dysplasia A: SNHL or mixed HL in 80% |
|
|
Twelve Craniofacial features of Down syndrome
|
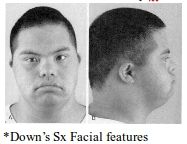
A: Brachycephaly/Flat occiput A: Small ears with Narrow EACs, low set A: Upslanting palpebral fissures A: Epicanthic folds, Brushfield spots on iris A: Midface hypoplasia, microgenia A: Small nose A: Narrow nasopharynx A: Large fissured lips A: Large fissured tongue A: Dental abnormalities A: Short neck A: Subglottic stenosis A: Small larynx A: Atlantoaxial instability & subluxation |
|
|
Downs Peds patient with OSA and pulmonary hypertension: Two treatments
|
A: T and A A: Bronchoscopy A: ?rapid maxillary expansion 3: Use smaller ET tube in Down’s patients |
|
|
Eight Reasons why Downs are susceptible to OSA |
A: Hypoplastic midface and cranium A: Narrow nasopharynx A: Macroglossia A: Muscular hypotonia A: Obesity A: Increased susceptibility to upper respiratory tract infections A: Small larynx A: SGS 3: UPPP may be useful in this patient population |
|
|
Treacher-Collins syndrome (mandibulofacial dysostosis) |
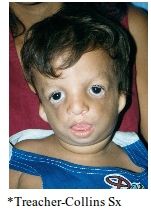
A: AD, 60% sporadic A: Mutation in TCOF1 gene, TREACLE protein, chromosome 5q A: Malformation of 1st & 2nd branchial arches A: Eye: Antimongoloid palpebral fissures (downslanting) Coloboma of the lower eyelids (upper lid in Goldenhar) Aplasia of lower lid lashes A: Ear: Microtia, , EAC stenosis or atresia, ossicular malformation, preauricular tags & fistulas, CHL in 30%, occasional SNHL (Mondini) A: Facial: Malar hypoplasia with non-fusion of zygomatic arches Hypoplastic supraorbital rims Flat nasofrontal angle Narrow nares, hypoplastic alar cartilages Tongues of hair onto cheeks A: Mandible & oral cavity: Mandibular hypoplasia (including condyle) Macrostomia High arched or cleft palate Dental abN A: May have choanal atresia A: Normal IQ |
|
|
Discuss Achondroplasia
|
A: Most common cause of short limb dwarfism A: AD, most sporadic, mutation of FGFR-3 gene, chromosome 4p16.3 A: Short limbs, genu varum, limited elbow extension, trident hand, long trunk, lumbar lordosis, frontal bossing, sunken nasal bridge, midface hypoplasia A: Normal cognition |
|
|
Apert (Acrocephalosyndactyly), Crouzon (Craniofacial dysostosis) and Pfeiffer syndromes |
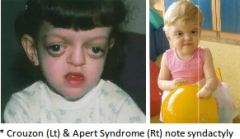
A: AD, mutations of FGFR-2 gene, chromosome 10q26 A: Craniosynostosis (Brachycephaly), midface hypoplasia, low nasal bridge, Parrotbeaked nose, choanal stenosis or atresia, mandibular prognathism, high arched palate, bifid uvula, cleft palate, and cervical fusion A: Hypertelorism, exophthalmos, and strabismus A: Cognitively normal to severe mental retardation A: Apert specific – Syndactyly, Stapes fixation (CHL) and patent Cochlear aqueduct A: Pfeiffer specific – Digital broadening |
|
|
Branchiootorenal syndrome (Melnick-Fraser syndrome)
|
A: AD, EYA1 gene, chromosome 8q A: Branchial cleft anomalies (63%) A: Otologic malformations – Hearing loss (89%), preauricular pits (77%), auricle abnormalities (41%), ossicular & cochlear malformations, lacrimal duct stenosis A: Renal dysplasia (66%) – Agenesis, polycystic kidneys, duplicated ureters 3: Renal abnormalities identifiable on IVP or renal U/S |
|
|
Goldenhar syndrome (oculoauriculovertebral spectrum) |
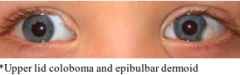
A: Most sporadic, some AD A: Unilateral facial asymmetry, Hemifacial microsomia A: Ocular – Upper lid coloboma, epibulbar dermoids A: Otologic – Mild deformity to Anotia, EAC atresia, ossicular abnormalities, CHL>SNHL A: Vertebral: Cervical fusion A: Others: Underdevelopment of Orbit, Facial muscles, Mandible |
|
|
Classification of hemifacial microsomia?
|
A: OMENS+ classification: O is for orbital distortion M is for mandibular hypoplasia E is for ear anomaly N is for nerve involvement S is for soft tissue deficiency Plus is used to include the expanded spectrum: cardiac, skeletal, pulmonary, renal, gastrointestinal, and limb anomalies. |
|
|
Maffucci syndrome |
A: Multiple Cavernous Hemangiomas, occasional visceral vascular lesions A: Dyschondroplasia & shortening/deformity of involved bones A: Chondrosarcoma in 25% |
|
|
Describe von Hippel Lindau syndrome (HIPPEL) |
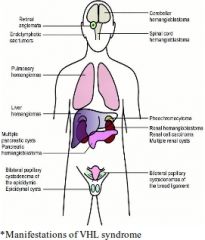
A: AD, mutation in the VHL gene, chromosome 3p25 A: Hemangioblastomas of CNS & retinas A: renal cysts/carcInoma A: Pheochromocytoma A: Pancreatic cysts A: Epididymal papillary cystademonata A: endoLymphatic sac tumors in 11% A: Dx criteria: Family history of von Hippel-Lindau (VHL) disease PLUS a tumour (CNS/retinal haemangioblastoma or clear cell renal cell carcinoma (RCC)); OR If no family history, ≥2 CNS/retinal haemangioblastomas plus visceral tumour (RCC, phaeochromocytoma or pancreatic tumour). |
|
|
Epidemiology of Choanal Atresia
|
A: Incidence 1:5000-8000 births A: F/M = 2/1 A: 50% have other anomalies (75% of bilateral cases) A: 60% mixed bony-membranous, 30% bony, 10% membranous A: 70% unilateral (60% of which are right-sided) |
|
|
Four methods of evaluating for Choanal Atresia |
A: Using cotton or mirror to detect airflow A: Inability to pass a small suction catheter A: Flexible scope A: CT scan |
|
|
Four parts to the anatomic deformity in Choanal Atresia |
A: Narrow nasal cavity A: Lateral bony obstruction from Pterygoid plate A: Medial bony obstruction from Vomer A: Membraneous obstruction |
|
|
General management approach of Choanal Atresia |
A: Unilateral – Nonurgent repair, ~1 year of age A: Bilateral – Establish airway & feeding pathway (McGovern nipple, Oropharyngeal airway; intubation not necessary unless mechanical ventilation required) A: Surgical repair approaches (SPAN = transSeptal, transPalatal, transAntral, transNasal) A: Postop care includes – ICU monitoring, frequent Suctioning, Antibiotics, PPI |
|
|
Syndromes are associated with Choanal Atresia (50% of all cases, CAT CTV) |
A: Crouzon syndrome A: Apert syndrome A: Treacher-Collins syndrome A: CHARGE syndrome A: Trisomies 18, 21 A: Velocardiofacial syndrome |
|
|
Describe CHARGE syndrome |
A: AD, CHD7 gene, chromosome 8q12 A: Coloboma A: Heart disease (endocardial cushion defect) A: Atresia (choanal) A: Retardation of growth, or mentation A: Genital defects (in males) A: Ear anomalies & deafness (CHL>SNHL) |
|
|
Embryologic spaces/structures of note in Glioma/Encephalocele formation |
A: Anterior neuropore A: Foramen Cecum (between frontal and ethmoid) A: Prenasal Space (between nasal bones and cartilaginous septum) A: Fonticulus Nasofrontalis (between frontal and nasal bones) |
|
|
Ddx of pediatric midline nasal mass |
A: Dermoid cyst (most common) A: Neurogenic – Glioma, Encephalocele, Neurofibroma A: Hemangioma |
|
|
Dermoid |
A: Epithelium lined, contains skin appendages, sinus tract leading to the skin A: Contains ectoderm and mesoderm A: Pathognomonic sign: Protruding hair (seen in a minority) A: Highly sensitive IC extension findings on imaging: Bifid crista galli and enlarged foramen caecum A: Dural connection in 30% |
|
|
Glioma |
A: Solid mass of Glial tissue with a fibrous stalk A: Dural connection in 15% A: 60% external (glabella), 30% internal (lateral nasal wall), 10% combined A: Path: dysplastic, neuroglial and fibrovascular tissue with NO ependymal tissue A: Manage any intracranial portion first; surgical excision through vertical midline dorsal excision, external rhinoplasty, or bicoronal approach |
|
|
Classification of congenital Encephaloceles |
A: Occipital – Most common, ~75% of cases A: Sincipital/frontoethmoidal, ~15% – nasoFrontal (most common subtype) nasoEthmoidal nasoOrbital A: Basal, ~10% – Transethmoidal (most common subtype) Sphenoethmoidal Transsphenoidal Sphenoorbital 3: Path: glial component with astrocytes surrounded by collagen, submucosal glands and sometimes septal cartilage with ependymal tissue (not present in gliomas) 3: Surgical excision needed within the first few months of life to minimize the risk of meningitis and cosmetic deformity open or endo |
|
|
Describe Furstenburg’s sign |
A: Expansion of a nasal mass with compression of the both IJV’s, associated with encephalocele, but not glioma or dermoid |
|
|
Parson’s major criteria (7) for chronic pediatric sinusitis |
A: Chonic nasal obstruction A: Nasal discharge A: Postnasal drainage A: Chronic cough A: Halitosis A: Headache A: Behavioral change |
|
|
How to diagnose peds acute ARBS according to AAP guidelines 2001? |
A: Infection of the paranasal sinuses lasting less than 30 days that presents with either persistent or severe symptoms A: Persistent symptoms are those that last longer than 10 to 14, but less than 30, days. Such symptoms include nasal or postnasal discharge (of any quality), daytime cough (which may be worse at night), or both. A: Severe symptoms include a temperature of at least 102°F (39C) and purulent nasal discharge present concurrently for at least 3 to 4 consecutive days in a child who seems ill. 3: Subacute 4-12 weeks, chronic > 12 weeks, recurrent acute bacterial sinusitis defined as having had 3 episodes in 6 months or 4 episodes in 12 months |
|
|
Abx in ABRS according to Antimicrobial guidelines for the treatment of ABRS in immunocompetent children, 2002? |
A: Abx given for 10-14 days. If no improvement in Sx after 72 hrs consider an alternative Abx. If pt. is NOT aSx after completing the course of Abx → cont. on Abx for 7-10 days A: Mild ARS & no Abx in the past 4-6 weeks: Amoxicillin (45–90 mg/kg per day), Amoxicillin/clavulanate (45–90 mg/kg per day), Cefpodoxime proxetil, Cefuroxime axetil, If allergic to β –lactams: TMP/SMX, Azithromycin, clarithromycin, or erythromycin. A: Mild ARS with Abx in the past 4-6 weeks OR Severe ARS with no Abx in the past 4-6 weeks: High-dose amoxicillin (90 mg/kg/day), Amoxicillin/clavulanate (high-dose amoxicillin component), Cefpodoxime proxetil or cefuroxime axetil. A: Severe ARS with Abx in the past 4-6 weeks: Amoxicillin/clavulanate or combination therapy (amoxicillin or clindamycin plus cefpodoxime or cefixime) |
|
|
Viruses most commonly associated with acute rhinosinusitis |
A: Rhinovirus A: Influenzae A: Parainfluenza A: Adenovirus 3: Others may include coronavirus, and RSV |
|
|
Bacteriology of acute pediatric sinusitis |
A: Streptococcus pneumonia A: Moraxella catarrhalis A: Haemphilus influenzae |
|
|
Bacteriology of chronic pediatric sinusitis |
A: Aerobes: S. pneumonia, M. catarrhalis, H. influenzae, S. aureus, α-hemolytic Strep, P. aeruginosa A: Anaerobes: Peptococcus, Peptostreptococcus, Bacteroides |
|
|
Indications for CT scanning for pediatric rhinosinusitis |
A: Severe illness or Toxic condition A: Immunocompromise A: Acute RS that does not improve with medical therapy in 48-72 hours A: Suppurative Complication |
|
|
Garcia & Harris indications of draining an orbital subperiosteal abscess? |
A: Age>9 A: Large size >10 mm A: Acute optic nerve or retinal compromise A: frontal sinusitis A: Non-medial subperiosteal abscess A: Chronic sinusitis A: Odontogenic source A: Suspicion of anaerobic subperiosteal infection (e.g., presence of gas within the abscess space as visualized on CT scan) A: Recurrent/prior I&D A: Others that are not included: Worsening despite medical Tx, lack of improvement in 48 hrs. |
|
|
5 indications for pediatric maxillary sinus aspirate |
A: Severe Toxic child A: Immunocompromise A: Unresolving symptoms after 72 hours A: Suppurative complications A: Work up for fever of unknown origin |
|
|
Absolute Indications for FESS in children |
A: Massive polyps in CF A: Antrochoanal polyp A: Fungal sinusitis A: Mucocele A: Intracranial complication A: Orbital abscess A: Traumatic injury to optic nerve A: dacrocystorhinitis due to sinusitis and resistant to medical Tx A: Meningoencephaloceles and neoplasms 3: Relative indication = CRS exacerbation despite maximal medical management |
|
|
Immune workup for recurrent sinusitis |
A: IgG subclasses A: IgM A: IgA A: IgE A: Ability to respond to polysaccharide antigens of S. pneumoniae, and H. flu |
|
|
Lab finding with common variable hypoglobulinemia |
A: Consistently low total immunoglobulins |
|
|
2 ways pediatric allergic fungal sinusitis is different from adult |
A: More likely to facial skeleton abnormalities A: More likely unilateral |
|
|
Bacteriology of pediatric Acute Sialadenitis |
A: Staphylococcus aureus A: Streptococcus viridans A: Streptococcus pneumoniae A: Streptococcus micros A: Esherichia coli A: Bacteroides melaninogenicus |
|
|
Epidemiology & classification of Cleft Lip and Palate |
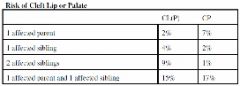
A: Second most common malformation after club foot A: CL +/- P in 1/1000 births, more in native Americans, M:F 2:1 A: Isolated CP occur in 1/2000 births, does NOT vary among ethnic groups, M:F 1:2 A: 70% of CL+/-P nonsyndromic, 50% of CP nonsyndromic A: Risk of inheritance in non syndromic CL&P and classification systems below (there are a few more) |
|
|
Three diagnostic signs of submucous cleft palate
|
A: Bifid uvula A: Muscular diastasis of the soft palate (zona pellucida) A: Notched hard palate |
|
|
Environmental factors contributing to cleft palate |
A: Drugs – Phenytoin, Thalidomide, Vitamin A derivatives, Folic acid antagonists, steroids in 1st trimester A: Smoking & Alcohol use in 1st trimester A: Amniotic band syndrome, maternal diabetes |
|
|
What is SIMONART’S BAND? |
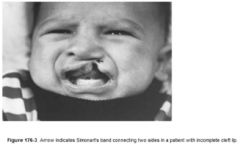
A: In an incomplete CL, bridge or bar of lip tissue of varying size that bridges the cleft gap |
|
|
Things to follow in Cleft L&P patients?
|
A: CLP team consultation A: Growth & Feeding: haberman/mead johnson/pigeon bottles A: Hearing screening & F/U A: SLP referral & F/U A: Genetic counselling A: Psych/social issues A: Orthodontic evaluation |
|
|
Lip adhesion |
A: If necessary, done @ 2-4 weeks with definitive repair at 4-6 months of age A: For unilateral, bilateral, or asymmetric wide complete cleft lip and palate |
|
|
Five options for Cleft Lip repair |
A: Rule of 10’s = 10 weeks, 10 pounds, 10 g of hemoglobin A: Straight line closure (rarely used anymore) A: Millard rotation advancement technique A: Millard bilateral cleft repair A: Tennison-Randall or Skoog techniques (single) triangular flap interdigitation A: Bardach (double) triangular flap interdigitation |
|
|
Cleft Palate repair |
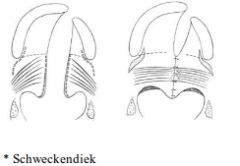
A: Performed at 5-15 months A: Restoration of soft palate sling incorporating tensor and levator palate muscles A: Soft palate repair only: Schweckendiek (primary veloplasty), obturate HP until older A: Secondary CP repair: If isolated 2ndry CP: Von Langenbeck (bipedicled flap palatoplasty) If primary + secondary CP: (but just fixing secondary P) Wardill-Kilner-Peet V-Y pushback technique Bardach uni-pedicle two flap palatoplasty A: Repair of complete cleft of primary & secondary palate or 2ndy cleft palate alone or SM cleft: Furlow double opposing Z-plasty |
|
|
Characteristics of the unilateral Cleft Lip Nasal deformity
|
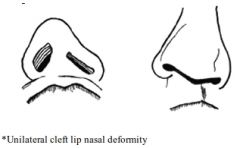
A: Nasal tip deflected to noncleft side with short medial crus and long lateral crus on cleft side A: Columella lies on noncleft side 2ndary to unopposed action of intact orbicularis oris A: Septum deflected to noncleft side A: Nasal dorsum to tilt to the cleft side due to underdeveloped nasal bones and nasal process of the maxilla A: Lat crus of LLC caudally displaced on cleft side A: Nostril on cleft side horizontally oriented (rather than vertical) A: Alar base on cleft side displaced lat/inf/post A: Deficiency of maxillary bone on cleft side (nasal floor often absent) 3: Bil. CL nasal deformity: lack of adequate columellar tissue (Alar cartilage & skin deficient), tip broad and flat, ala laterally displaced with horizontal oriented nostrils |
|
|
Outline the timeline of Cleft Lip and Palate repair
|
A: 3 months – Cleft lip repair, PET insertion A: 1 year – Cleft palate repair A: 5 years – Columellar lengthening (for bilateral cleft lip) A: 8-16 years – Orthodontics A: 10 years – Alveolar cancellous bone graft A: 14 years – Definitive rhinoplasty and orthognatic surgery |
|
|
List five syndromes associated with Cleft Palate (TDP CAVS) |
A: Stickler A: Treacher-Collins A: Pierre-Robin sequence A: Apert A: Velocardiofacial A: Down A: Crouzon’s |
|
|
Passavant’s ridge |
A: Prominence on the posterior wall of the nasopharynx from contraction of the superior constrictor during swallowing |
|
|
Management of velopharyngeal insufficiency (VPI) |
A: Non-surgical – Speech therapy, Prosthetics (palatal lift or obturator), Biofeedback with nasometry A: Surgical – Pharyngoplasty, Pharyngeal flaps, Posterior pharyngeal wall augmentation |
|
|
Name all 6 components of Walderyer’s ring |
A: Lingual tonsils A: Palatine tonsils A: Adenoid A: Gerlach’s tonsils (Tubal, posterior to ET opening) A: Lateral bands A: Posterior pharyngeal wall |
|
|
Adenoid blood supply (5) |
A: Pharyngeal branch of the internal maxillary (major supply) A: Ascending pharyngeal artery A: Ascending palatine branch of the facial artery A: Ascending cervical branch of thyrocervical trunk A: Artery of the pterygoid canal |
|
|
Adenoid innervation |
A: CNs IX & X |
|
|
Adenoid histology |
A: Ciliated pseudostratified columnar epithelium A: Stratified squamous epithelium A: Transitional epithelium 3: Inflammation increases specialized squamous epithelium proportion and decreases respiratory proportion |
|
|
Four zones of antigen processing in adenotonsillar tissue |
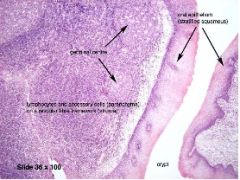
A: Specialized squamous epithelium (Dendritic cells) A: Extrafollicular area (T-cells) A: Mantle zone of lymphoid follicle (Mature B-lymphocytes) A: Germinal center of follicle (Active B-cells) |
|
|
Tonsil blood supply
|
A: Lower pole: Facial artery (Tonsillar & Ascending Palatine branches) Dorsal lingual A: Upper pole: Internal maxillary artery (lesser +/- greater palatine art) Ascending pharyngeal artery |
|
|
Describe efferent lymphatics of the tonsils and adenoids |
A: Upper deep cervical nodes (T+A) A: Retropharyngeal LN (A only) 3: No afferent lymphatics for T and A |
|
|
Major immunologic product of tonsils and adenoids |
A: Secretory IgA |
|
|
Four symptoms of Acute Adenoiditis |
A: Purulent rhinorrhea A: Nasal obstruction A: Fever A: Otitis media |
|
|
Recurrent acute adenoiditis |
A: 4+ episodes in a 6 month period |
|
|
Four symptoms of Chronic Adenoiditis |
A: Persistent nasal discharge A: Chronic congestion A: Postnasal drip A: Halitosis |
|
|
Obstructive adenoid hyperplasia triad |
A: Chronic nasal obstruction (obligate mouth breathing, snoring) A: Rhinorrhea A: Hyponasal speech |
|
|
Five symptoms of Acute Tonsillitis |
A: Erythematous exudative tonsils A: Sore throat A: Dysphagia A: Tender cervical adenopathy A: Fever |
|
|
Recurrent Acute Tonsillitis |
A: 7 infections in a year A: 5 infections for 2 consecutive years A: 3 infections for 3 consecutive years |
|
|
Five symptoms/signs of Chronic Tonsillitis |
A: Chronic sore throat A: Halitosis A: Tonsilloliths A: Peritonsillar erythema A: Persistent tender cervical adenopathy |
|
|
Four symptoms/signs of Obstructive Tonsillar hyperplasia |
A: Enlarged tonsils A: Snoring A: Obstructive disturbances A: Dysphagia and voice changes |
|
|
Seven Etiologies of pseudomembranous tonsillitis |
A: Epstein-Barr virus (mononucleosis) A: Group A ß-hemolytic Streptococcus A: Corynebacterium diphtheriae A: Neisseria gonnorheae A: Syphilis A: Candidiasis A: Vincent’s angina |
|
|
Define Centor criteria for likelihood of acute GABHS tonsillitis? |
A: Four criteria: History of fever Tonsillar exudates Tender anterior cervical adenopathy Absence of cough A: Each criteria = 1 point A: Management: 0-1 points - No antibiotic or throat culture necessary (Risk of strep. infection <10%) 2-3 points - Throat culture and treat with an antibiotic if culture is positive (Risk of strep. infection 32% if 3 criteria, 15% if 2) 4 points - Treat with antibiotic (Risk of strep. infection 56%) |
|
|
Indications for Adenoidectomy (ION) |
A: Infection – Recurrent acute/chronic Adenoiditis, recurrent acute/chronic Otitis Media with or without Effusion (kids >4 years benefit from adenoidectomy with the Second set of PETs), chronic Sinusitis A: Obstruction – Adenoid hyperplasia with chronic Nasal Obstruction or obligate Mouth Breathing, Sleep-related disordered breathing (OSAS, OSHS, UARS), Cor pulmonale, FTT, Orofacial growth/dental/speech/swallowing abnormalities A: Neoplasia – Suspected, benign or malignant, Lymphoproliferative disorder 3: Absolute indications for adenoidectomy (Darrow, 2002) OSA Suspected Malignancy FTT Abnormal dentofacial growth Relative: UAO (SDB) Dysphagia Speech impairment Halitosis Otitis media Recurrent or chronic rhinosinusitis or adenoiditis Clinical indicators for adenoidectomy as recommended by the AAO-HNS in 2000 are: Four or more episodes of recurrent purulent rhinorrhea in prior 12 months in a child <12. One episode documented by intranasal examination or diagnostic imaging. Persisting symptoms of adenoiditis after 2 courses of antibiotic therapy. One course of antibiotics should be with a betalactamase stable antibiotic for at least 2 weeks. Sleep disturbance with nasal airway obstruction persisting for at least 3 months. Hyponasal or nasal speech Otitis media with effusion >3 months or second set of tubes Dental malocclusion or orofacial growth disturbance documented by orthodontist. Cardiopulmonary complications including cor pulmonale, pulmonary hypertension, and right ventricular hypertrophy associated with upper airway obstruction. Otitis media with effusion over age 4. |
|
|
Four contraindications for Adenoidectomy |
A: Cleft palate A: Submucous cleft A: Hypernasal speech A: Nasal regurgitation 3: All are signs of VPI |
|
|
Risk factors for VPI after adenoidectomy |
A: History of nasal fluid regurgitation A: Occult submucus cleft A: Family history of clefts A: Neuromuscular problems (CNS) |
|
|
Indications for Tonsillectomy (ION) |
A: Infection – Recurrent acute/chronic Tonsillitis, Halitosis, Complications (Cervical Abscess, Airway obstruction Cardiac valve disease, recurrent febrile Seizures, tonsillar Hemorrhage), streptococcus Carrier unresponsive to medical treatment, recurrent or unresponsive Peritonsillar Abscess A: Obstruction – Tonsillar hyperplasia with obstruction, Sleeprelated disordered breathing (OSAS, OSHS, UARS), Cor pulmonale, FTT, Orofacial growth/dental/speech/swallowing abnormalities A: Neoplasia – Suspected, benign or malignant, Lymphoproliferative disorder 3: Absolute indications for tonsillectomy (Darrow, 2002) Suspected Malignancy FTT OSA Abnormal dentofacial growth hemorrhagic tonsillitis Relative: UAO (SDB) Dysphagia Speech impairment Halitosis Recurrent or chronic pharyngotonsillitis, peritonsillar abscess Streptococcal carriage Clinical indicators for tonsillectomy as recommended by the AAOHNS in 2000 are: Patient with 7 or more episodes of tonsillitis in the preceding year, OR 5 or more episodes in each of the preceding 2 y, OR 3 or more episodes in each of the preceding 3 y despite adequate medical therapy (with documentation in the medical record for each episode of sore throat and one or more of the following: temperature >38.3°C, cervical adenopathy, tonsillar exudate, or positive test for GABHS) → Paradise criteria Hypertrophy causing dental malocclusion or adversely affecting orofacial growth documented by orthodontist. Hypertrophy causing upper airway obstruction, severe dysphagia, sleep disorders, or cardiopulmonary complications. Peritonsillar abscess unresponsive to medical management and drainage documented by surgeon, unless surgery performed during acute stage. Persistent foul taste or breath due to chronic tonsillitis not responsive to medical therapy. Chronic or recurrent tonsillitis associated with the streptococcal carrier state and not responding to beta-lactamase resistant antibiotics. Unilateral tonsil hypertrophy presumed neoplastic |
|
|
Four contraindications for Tonsillectomy (LASH CPA) |
A: Leukemias A: Agranulocytosis A: Systemic disease that is uncontrolled (DM, TB) A: Hemophilias 3: Relative contraindications = Cleft Palate, and Acute infection |
|
|
Eight Criteria which should suggest an overnight stay posttonsillectomy |
A: Under 3 years of age A: OSA A: Craniofacial abnormalities A: Neuromuscular dz – CP, duchenne, down’s A: Mucopolysaccharidosis – hunter’s, hurler’s A: Medical comorbidity (diabetes, seizures, asthma, cardiac disease, etc) A: Peritonsillar abscess A: Emesis or hemorrhage A: Patient lives greater than 60 minutes away from hospital A: Poor socioeconomic class predisposing to neglect 3: Indications for overnight monitoring according to CPG 2011: Age <3 and severe OSA (AHI>10 or O2 sat nadir <80%) |
|
|
Indications for PSG prior to Tonsillectomy (CPG 2011)? |
A: Need for surgery is uncertain A: When there is discordance between tonsillar size on PE and the reported severity of SDB A: Obesity A: Down syndrome A: Craniofacial abnormalities A: Neuromuscular disorders A: Sickle cell disease A: Mucopolysaccharidoses |
|
|
Complications of Adenotonsillectomy |
A: Halitosis (most common) A: Dehydration A: Postoperative hemorrhage – 0.5-1% A: Pulmonary – postoperative edema & hypoxemia (loss of auto- PEEP, hypercapneic respiratory drive) A: Airway obstruction A: Velopharyngeal insufficiency A: Nasopharyngeal stenosis A: Atlantoaxial subluxation (Grisel’s syndrome) – Down syndrome at higher risk A: Eagle syndrome A: Death – 1/25,000 |
|
|
Six reasons for post-adenotonsillectomy desaturation |
A: Post-obstructive pulmonary edema A: Loss of hypercapnic respiratory drive A: Airway swelling & obstruction A: Aspiration of blood clots A: Laryngospam A: Narcotic overmedication |
|
|
Define Grisel’s syndrome |
A: Alanto-axial joint laxity due to adenoidectomy A: Vertebral body decalcification and laxity of anterior transverse ligament between axis and atlas due to inflammation/infection in the nasopharynx A: Spontaneous subluxation occurs 1 week post op – Pain and Torticollis |
|
|
Management of Peritonsillar Abscess (infection of a peritonsillar salivary/Weber gland) |
A: Hydration A: Analgesia A: Incision & drainage or needle aspiration (75% effective), culture A: Antibiotics, parenteral, enteral, or combined A: If unsuccessful – CT scan, reincision & drainage, Quinsy tonsillectomy |
|
|
Six Complications of Peritonsillar Abscess |
A: Airway obstruction A: Dehydration A: Spread to other spaces A: Carotid artery erosion A: IJV thrombophlebitis (Lemierre’s syndrome) A: Sepsis |
|
|
Causes of Unilateral Tonsillar hyperplasia |
A: Neoplastic – Lymphoma A: Infectious – Mycobacteria (tuberculosis, atypical), Actinomycosis, Fungal |
|
|
Ddx of a Congenital Tonsillar mass (3) |
A: Teratoma A: Hemangioma A: Lymphatic malformation |
|
|
High risk groups for Pediatric OSAS |
A: Abnormal airway anatomy – Down’s, Pierre-Robin, Achondroplasia, Craniosynostoses, Treacher-Collins, Macroglossia, Klippel-Feil, Pyriform aperture stenosis, Laryngomalacia, Masses A: Neuromuscular disease – CP, Down, Hypothyroidism, Muscular dystrophies, Seizures, Prader-Willi, Chiari malformations A: Cardiovascular disease – Pulmonary/systemic HTN, Congenital heart disease A: Morbid obesity A: Pectus excavatum/Scoliosis |
|
|
Ten Differences in adult vs pediatric OSAS |
A: Snoring intermittent vs continuous A: Mouth breathing rare vs common A: Obesity common vs rare A: Failure to thrive & enuresis rare vs common A: Daytime somnolence common vs rare A: Hyperactivity, attention deficit, aggression rare vs common A: Nighttime arousals common vs rare A: Gender predilection male vs none A: CPAP mainstay vs selected (postop OSA, craniofacial anomaly, C/I to surgery) A: Surgery selected vs mainstay |
|
|
Criteria for abnormal polysomnogram in pediatric OSAS |
A: Apnea index ≥1 (lasting longer than two consecutive breaths) A: Oxygen desaturation to <92%, >4% x >3 times/hour, or associated with a change in heart rate >25% A: End tidal CO2 >50mmHg >8% of total sleep time (or >53 at any point), or >45mmHg >60% of total sleep time |
|
|
Indications for PSG postsurgery for pediatric OSAS |
A: Persistent snoring A: Preoperative Severe OSA A: Preoperative OSA complications A: Age <1 year |
|
|
Hunter’s syndrome |
A: X-linked, type II mucopolysaccharidosis A: Deficiency of beta-galactosidase A: Macrocephaly, broad face, low nasal bridge, death usually occurs from infiltrative CMO & valvular disease leading to CHF |
|
|
Hurler’s syndrome |
A: AR, Type 1 mucopolysaccharidosis A: Deficiency of α-1-iduronidase which breaks down heparan-, dermatan- and keratan-sulfates A: Visceromegaly, macroglossia, macrocephaly, progressive neurologic dysfunction, death in 1st decade |
|
|
Differences between the pediatric & adult larynx |
A: Pediatric larynx higher in neck (C3/4 vs C5/6) A: Epiglottis curved/omega shaped, in contact with soft palate A: Thyroid cartilage oblique, no defining angle, overlapping with hyoid & cricoid A: Arytenoids are relatively large A: Infant vocal cords 4-4.5mm long at birth, adults 14-23mm A: Infant true vocal cord 50% composed of vocal process of arytenoid, in adults 25-33% A: Funnel shape, Infant subglottis narrowest portion of airway, 4.5-5 mm at full term A: Infant subglottis is loose, with a lot of submucosal glands |
|
|
Age when cricoid is no longer narrowest segment of airway |
A: 8 years |
|
|
Embryology of the tracheoesophageal system |
A: During the fourth week (26 days) A: Laryngotracheal groove appears as median outgrowth from the caudal end of the ventral wall of the primitive pharynx A: Laryngotracheal diverticulum forms by 28 days A: Tracheoesophageal folds fuse into a septum that separates the laryngotracheal tube A: Arytenoid swellings from neural crest cell-derived mesenchyme of the 4th and 6th arches A: Laryngeal epithelium occludes lumen at 8th week, and recanalization occurs by 10th week. |
|
|
Prenatal sonographic findings of CHAOS? |
A: Increased in lung size and echogenicity A: Fluid-filled & dilated trachea A: Fetal hydrops A: Polyhydramnios |
|
|
Location of stridor by its pattern |
A: Inspiratory = dynamic supraglottis and glottis A: Biphasic = subglottis and cervical trachea A: Expiratory = fixed intrathoracic trachea |
|
|
Stridor history mnemonic “SPEC SPEAR” |
A: S = Severity, parents’ Subjective impression A: P = Progression of obstruction over time A: E = Eating/feeding difficulties A: C = Cyanotic spells A: S = Sleep disordered breathing A: P = history of Prematurity A: E = history of Endotracheal intubation A: A = possibility of foreign body Aspiration A: R = Radiographics that detect a specific abnormality |
|
|
10 Signs of airway obstruction |
A: Stridor A: Dyspnea, tachypnea A: Tachycardia A: Diaphoresis, circumoral pallor, anxiety/restlessness A: Retractions – tracheal tug, suprasternal, intercostal, substernal A: Flaring of nasal alae, A: Use of accessory respiratory muscles A: Cyanosis, in extreme cases A: Respiratory arrest |
|
|
The 5 “A” of stridor |
A: Age A: Acuteness A: Appearance (toxic or non-toxic) A: Acoustics (volume, pitch, phase) A: Associated symptoms (dysphonia, cough, drooling, posturing, dysphagia) |
|
|
Airway imaging modalities |
A: Plain soft tissue films of the neck, AP (croup) + lateral (epiglottittis & RPA) A: CXR AP + lateral (FB & tracheal stenosis) A: Inspiratory & expiratory chest films (FB) A: Airway Fluoroscopy (dynamic, awake & sleep, best for OSA) A: Barium swallow (vascular compression) A: Electron beam CT, or spiral CT scan with apnea A: MRI of the airway (intrathoracic vascular anomalies & masses) A: Bronchogram (after MRI, if difficult tracheobronchial stenosis) A: Laryngeal U/S (linear 7.0, 10 MHz, B-mode real-time) |
|
|
Indications for endoscopic examination in pediatric stridor (PAUSe) |
A: Progressive stridor A: parental Anxiety A: Unusual features – Cyanotic attacks, apneic attacks, dysphagia, aspiration, recurrent pneumonias, failure to thrive, radiologic abnormality A: Severe stridor |
|
|
What 4 pediatric airway abnormalities are improved in the prone position? |
A: Laryngomalacia A: Pierre-Robin sequence A: Vascular compression A: Mediastinal mass |
|
|
Clinical characteristics of Laryngomalacia |
A: Variable inspiratory stridor – Begins in first few days/weeks after birth, worse with crying, feeding, or supine position with H&N flexed, better when prone or with H&N extended A: Signs of intermittent upper airway obstruction A: Normal cry A: Normal general health and development |
|
|
Factors influencing development of laryngomalacia |
A: Shortened aryepiglottic folds A: Anterior collapse of cuneiform cartilage A: Immature neuromuscular control A: Reflux |
|
|
How is LPR different from classic GERD? |
A: Patients have head & neck symptoms but heartburn is uncommon A: Predominantly upright (day-time) reflux A: Normal esophageal motility A: Most do not have esophagitis, as in GERD A: Laryngopharyngeal epithelium is more susceptible to reflux related injury than esophageal epithelium |
|
|
Six Evaluations for Laryngopharyngeal Reflux (LPR) |
A: Gastric emptying scan (milk scan) technetium 99 m A: 24 hour double-probe pH monitoring A: Barium swallow A: Broncho-alveolar lavage for lipid laden macrophages (LLM, 70% needed) A: Esophagogastroduodenoscopy with Biopsy (EGD, suspected eosinophilic esophagitis) A: Diagnostic markers pepsin & carbonic anhydrase isoenzyme III (CA-III) |
|
|
Non-surgical management of laryngomalacia |
A: Observation (90%) A: Anti-reflux A: Position A: Thickened feeds A: Frequent smaller meals A: CPAP |
|
|
Five Indications for surgeries for severe laryngomalacia (10%) |
A: FTT, weight loss, feeding difficulty A: Life-threatening episode, cyanotic attacks, respiratory distress, documented desaturation A: Cor pulmonale, pulmonary hypertension A: Obstructive sleep apnea A: Severe chest deformity |
|
|
Interventions for severe laryngomalacia |
A: Supraglottoplasty/epiglottoplasty – Unilateral or bilateral, division of aryepiglottic fold, partial epiglottis amputation, removal of redundant supra-arytenoid mucosa and lateral borders of epiglottis, removal of cuneiform & corniculate cartilages A: Epiglottopexy with glossoepiglottic adhesion A: Tracheostomy (rare) |
|
|
Six Complications of Supraglottoplasty |
A: Bleeding A: Temporary dysphagia A: Aspiration A: Temporary worsening of airway A: Supraglottic Stenosis (< 5% if bilateral, preserve islands of mucosa, esp. interarytenoid) A: Re-operation (15% if unilateral) |
|
|
Methods of Voice Assessment |
A: Audiotape & videotape Recording (speech therapist) A: Parent & child Questionnaire A: Fiberoptic laryngoscopy with video-stroboscopy A: Spectral voice Analysis = Multi-Dimensional Voice Program (MDVP) |
|
|
Ddx of pediatric vocal cord paralysis (NATIVE) |
A: Neurologic (brainstem tumor, CP, hydrocephalus, meningomyelocele, hypoxic encephalopathy, hypotonia) A: Arnold-Chiari malformation (ALWAYS consider in neonate) A: Birth Trauma (C-section, nuchal cord, Recovery >9 mos) A: Iatrogenic injury (CVS, PDA, TEF) A: Idiopathic (47%, especially bilateral) A: Infectious – Syphilis A: Vascular anomalies A: Everything else – Mobius, Charcot-Marie Tooth 3: >50% Bilateral |
|
|
4 types of Chiari malformations |
A: I – Protrusion of cerebellar Tonsils A: II – Protrusion of cerebellar Vermis, lower Pons and Medulla (Arnold-Chiari malformation) A: III – Herniation of Cerebellum (high cervical meningocele) A: IV – Cerebellar Hypoplasia (Dandy-Walker syndrome) |
|
|
ENT manifestations of Arnold-Chiari malformation |
A: CN IX-XII difficulties A: Bilateral vocal cord paralysis, respiratory distress A: Poor Feeding A: Aspiration |
|
|
Management of Bilateral VCP |
A: +/- urgent intubation A: +/- ACLS management A: Supportive measures (upright positioning, thickening of formula, observation, management) A: Tracheotomy (> 2 years to allow for spontaneous recovery, 50%) A: Lateralization (arytenoidectomy +/- CO2 laser cordotomy, arytenoidopexy, cordotomy) A: Open procedures / Reanimation / Electrical pacers 3: Decannulation rates > 60% |
|
|
Benjamin & Inglis classification of Laryngeal Clefts |
A: I – Interarytenoid A: II – Partial cricoid (below VCs) A: III – Through entire cricoid into cervical trachea A: IV – Distal (thoracic) trachea |
|
|
Laryngeal cleft associated syndrome? |
A: Pallister-Hall Sx |
|
|
Best imaging study for Laryngeal Cleft |
A: Barium swallow (gastrograffin bad for lungs) |
|
|
Management of Laryngeal Clefts by grade |
A: I – Observation, anti-reflux & thickened feeds, vs. Endoscopic repair (2 layer closure) A: II or III – Laryngofissure for precise multilayered anatomic closure A: IV – Laryngofissure & Sternotomy vs. Lateral pharyngotomy & Thoracotomy (postop ECMO, no ETT) |
|
|
Anomalies associated with Laryngeal Clefts |
A: Esophageal atresia +/- TEF (#1, 6% of TEFs have clefts, continued aspiration) A: Cleft lip/palate A: CHD, GI anomalies A: G syndrome (Opitz-Friass) – Hypertelorism, Hypospadias, Cleft lip/palate A: Pallister-Hall syndrome – Bifid epiglottis, Hypothalamic hamartoblastoma, Hypopituitarism, Imperforate anus, Postaxial polydactyly |
|
|
Relative frequency of Laryngeal Webs by location |
A: Glottic = 75% A: Subglottic = 7% A: Supraglottic = 2% |
|
|
Investigations for Laryngeal Web |
A: Flexible laryngoscopy A: +/- Rigid laryngoscopy + bronchoscopy (site, thickness, horizontal and vertical extent) A: +/- X-ray (Sail sign = Persistent tissue between VCs + subglottis, R/O SGS) A: +/- FISH for anterior glottic web (22q11.2 deletion) |
|
|
Cohen’s classification of congenital glottic webs? |
A: Type I: It involves less than 35% of the glottis, no subglottic extension. A: Type II: The web involves 35-50% of the glottis and can be thin or thick, these may be associated with some subglottic extension of stenosis. A: Type III: This type involves 50-75% of the glottis. The web is usually very thick anteriorly and may thin out as it extends posteriorly; these almost always have a subglottic component to them. Patients have marked vocal dysfunction and have moderate-to-severe airway symptoms. A: Type IV: The web involves 75-90% or more of the glottis and is uniformly thick both anteriorly and posteriorly. The patient is usually aphonic. Severe airway obstruction is usually present and almost always requires an emergency tracheotomy. |
|
|
Treatment of Glottic Webs by type |
A: Observation, if possible delay surgery to >3 yo A: Endoscopic repair – Lysis via laser/cold knife, can be staged (one side then other), serial Dilations, +/- Stent, Suturing of free edges, Local flaps, Mitomycin C A: Type I – Divide with laser/cold steel at age 3-4 A: Type II – Incise along one cord then serial dilations or incise along other cord 2 weeks later; If keel required, trach needed A: Type III/IV – Trach, corrective procedure at age 3-4, Laryngofissure with Stent/Keel or LTR |
|
|
Laryngeal atresia types |
A: Complete absence of laryngeal lumen A: I – Supraglottic + Infraglottic A: II – Infraglottic A: III – Glottic |
|
|
Cri-du-Chat syndrome |

A: Deletion chromosome 5p (1/50 000 births) A: CNS – Microcephaly, 1% profound MR, hypotonia, CVD? A: ENT – High pitched stridor (cat cry), Hypertelorism, Broad nasal root, Cleft lip/palate A: Laryngoscopy – Elongated/narrowed diamond-shaped endolarynx, interarytenoid muscle paralysis, Cleft? |
|
|
Beckwith-Wiedemann syndrome
|
A: Sporadic occurrence A: “Overall growth syndrome”, Macroglossia, Omphalocele, Visceromegaly, Cytomegaly of adrenal cortex A: Macroglossia may cause airway obstruction or chronic alveolar hypoventilation |
|
|
Ddx of small blue cell malignancies of childhood (5)
|
A: Rhabdomyosarcoma A: Undifferentiated soft-tissue sarcomas A: Lymphoma A: Primitive NeuroEndocrine Tumor (PNET)/Ewing’s sarcoma A: Neuroblastoma A: Others: Melanoma/Merckel cell Ca/SNUC/plasmacytoma in adults |
|
|
Ddx of aural polyp in pediatrics |
A: Cholesteatoma A: Eosinophilic granuloma (Type 1 LCH) A: Rhabdomyosarcoma |
|
|
Most common benign neoplasm of larynx in children |
A: Recurrent respiratory papillomatosis (RRP) 3: Second most common cause of hoarseness in children |
|
|
Etiology and types of RRP |
A: Etiology – HPV infection (6 & 11) A: Juvenile onset (<12 years) – More common & more aggressive, typically diagnosed by 2-4 years, vertical transmission from mother during parturition (risk 1:80-1:500) A: Adult onset (>12 years) – Peaks between 20-40 years |
|
|
Triad of symptoms for RRP |
A: Progressive hoarseness (most common symptom) A: Stridor A: Respiratory distress |
|
|
Eight Common sites of lesion for RRP |
A: Limen Vestibuli (junction of the nasal vestibule and nasal cavity proper) A: Nasopharyngeal surface of Soft Palate A: Midline of laryngeal surface of Epiglottis A: Upper & lower Ventricle margins A: Undersurface of the Vocal Cords A: Carina A: Bronchial Spurs A: Tracheostomy site (iatrogenic junction) 3: Preferentially junction of squamous epithelium with respiratory epithelium |
|
|
Treatment modalities for RRP |
A: Tracheostomy – Increased risk of distal spread, avoid unless absolutely necessary A: Surgical debulking – Standard of care, remove as much disease as possible, preserve normal structures A: Adjuvant therapy – 10% of patients will require some form |
|
|
Possible surgical modalities for RRP |
A: Phono-microsurgery A: Microlaryngoscopy with Cup forceps A: Microlaryngoscopy with Microdebrider A: Microlaryngoscopy with Laser (KTP, Nd:YAG, CO2, flash lamp pulsed dye) A: Cryosurgery |
|
|
10 adjuvant therapies for RRP |
A: a-Interferon therapy (most common, exact mechanism unknown) A: Indole-3-carbinol diet supplementation (found in cruciferous vegetables) A: Retinoic acid A: Methotrexate A: Antivirals (Ribavirin, Acyclovir, Cidofovir) A: HspE7 (in phase 2 trials currently) = Heat shock protein to E7 A: Photodynamic therapy A: Mumps vaccine |
|
|
Criteria for use of adjuvant therapies for RRP |
A: >4 surgeries/year A: Rapid regrowth with airway compromise A: Distal multisite spread of disease |
|
|
Triad of juvenile onset RRP risk factors |
A: First born (75% of cases) A: Vaginal delivery A: Teenage mother A: other include prolonged 2nd stage of labor |
|
|
Adult onset RRP risk factors |
A: Frequent oral sex >6 A: Multiple sex partners >26 |
|
|
RRP prophylactic Cesarean section recommendations |
A: Not routinely recommended A: Should be strongly considered in Young, Primiparous mothers with recent HPV infection and genital warts A: ~1:400 risk of vertical transmission of HPV in mother with condyloma accuminata |
|
|
Three Risk factors for increased spread of RRP |
A: Juvenile A: Young age A: HPV type 11 |
|
|
Percentage of extralaryngeal spread of adult and juvenile onset RRP |
A: 30% Children A: 15% Adult |
|
|
Natural history of RRP |
A: Spontaneous regression A: Not associated with puberty |
|
|
Seven most common GER related laryngeal disorders in pediatrics (CHARLES) |
A: Chronic Cough A: Hoarseness A: Aspiration A: Recurrent Croup A: Laryngomalacia A: Episodic Laryngospasm A: Subglottic Stenosis |
|
|
Four anatomic structures are seen on Barium swallow causing compression |
A: Cricopharyngeus (C6) A: Aortic arch (T4) A: Left mainstem bronchus (T6) A: Lower esophageal sphincter 3: Common sites of injury in caustic ingestion |
|
|
Seven most common congenital Vascular anomalies that can cause Tracheomalacia? |
A: Innominate artery compression (most common, arises more medially than normal, anterior compression) A: Double Aortic arch (most common true vascular ring): compresses both trachea and esophagus A: Right Aortic arch with left ligamentum arteriosum A: Anomalous left Carotid artery A: Anomalous right Subclavian artery (Dysphagia lusoria: arises from left descending aorta, passes behind E (85%) or between T&E (15%), associated with a non-recurrent right RLN) A: Pulmonary artery sling (left pulmonary artery arises off the right pulmonary artery, passes between T&E; associated with complete tracheal rings) A: Pulmonary artery dilation 3: 2 aortic, 2 pulmonary, 1 carotid, 1 innominate, and 1 subclavian |
|
|
Innominate artery compression pattern and specific sign to look for |
A: Compresses anterior tracheal wall (R more than L) A: Pulsatile, should affect the R arm pulses??? |
|
|
3 Absolute and 4 relative indications for surgical treatment for compression due to vascular anomalies (AFP RESP) |
A: Absolute – Reflex Apnea (from vagal stimulation) A: Absolute – Failure of medical management of Severe respiratory distress after 48 hours A: Absolute – Prolonged intubation A: Relative – Repeated URTI episodes A: Relative – Exercise intolerance A: Relative – Significant FTT & dysphagia A: Relative – Coexisting Pathology (SGS, asthma, CF, or previous TEF repair) |
|
|
Clinical signs of post-obstructive pulmonary edema |
A: Pink frothy secretions A: Hypoxemia A: Bilateral end expiratory wheezing with rales A: Radiographic findings (increased pulmonary markings & fluid overload) |
|
|
Management of post-obstructive pulmonary edema |
A: Fluid restriction A: Diuretics A: CPAP |
|
|
5 TEF types in descending order of frequency (CAEBD) |
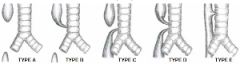
A: C:Esophageal atresia (EA) & distal TEF (85%) A: A: Isolated EA (10%) A: E: Isolated true TEF (H-type, 4%) A: B: EA with proximal TEF (0.5-1%) A: D: EA with double TEF (0.5) |
|
|
Presentation, 4 investigations, and 2 interventions for TEF
|
A: Present with immediate feeding problems and aspiration A: Investigations – Inability to pass a feeding tube, X-rays (curled tube in the pharynx & air bubble in the stomach), Fluoroscopy, Endoscopy (mainstay) A: Management – Surgical correction, Dilation of strictures |
|
|
Describe the components of VACTERL syndrome? |
A: V – Vertebral/Vascular anomalies A: A – Anal atresia A: C – Cardiac anomalies (PDA, valve problems) A: TE – Tracheoesophageal fistula A: R – Renal/Radial bone anomalies A: L – Limb anomalies (extra digits, shortened limbs) |
|
|
Eight tracheal causes of stridor |
A: Tracheomalacia A: Hemangioma A: Vascular compression A: TE fistula A: Tracheal stenosis A: Tracheal cyst A: Tracheal agenesis (with a TEF) A: Tracheal bronchus |
|
|
Nine Surgical options of Tracheal Stenosis |
A: Endoscopic (Balloon or serial dilatations, Cold knife, Laser, Stents) A: Augmentative (Tracheoplasty with Rib cartilage, Pericardium) A: Resection and Anastomosis (Wedge resection, Segmental resection <50% tracheal length, Slide tracheoplasty) A: Cadaveric Homograft with Dumon stent |
|
|
8 methods of reducing tension across the Tracheal Anastomosis suture line |
A: Suprahyoid release A: Infrahyoid release A: Peritracheal mobilization A: Intercartilaginous incisions A: Perihilar release A: Dissection of the pulmonary vasculature A: Transplantation of the left mainstem bronchus A: Neck flexion with chin-to-chest suturing(Grillo/Guardian stitch) A: How much tension is too much? Apparently 1700 gm. How do you measure? No clue. Any clinical significance? I don’t think so. |
|
|
Five complications of tracheal stenosis repair |
A: RLN damage A: Granulation tissue A: Infection A: Dehiscence A: Fistulization |
|
|
Agents responsible for caustic ingestion and their damage pattern |
A: Corrosives/Acids – Coagulative necrosis, superficial coagulum prevents deeper damage A: Caustics/Bases – Liquefactive necrosis, early disintegration of tissues with deep penetration, significant damage at pH >12 A: Bleaches – pH ~7, esophageal irritants |
|
|
Three most important questions on history about Caustic agents |
A: pH A: Volume ingested A: Consistency |
|
|
Staging of Esophageal Burn injury |
A: Grade 1 – mucosal erythema (superficial) A: Grade 2 – mucosal erythema with noncircumferential exudate (transmucosal) A: Grade 3 – circumferential exudates (transmucosal) A: Grade 4 – circumferential exudates with perforation (transmural) |
|
|
Three degrees of Endoscopic Appearance for esophageal burn injury |
A: First Degree – Nonulcerative esophagitis, mild erythema, edema of mucosa A: Second Degree – Whitish exudate, erythema, underlying ulceration that may extend into the muscularis A: Third Degree – Dusky or blackened transmural tissue, deep ulcerations that may extend into periesophageal tissue, lumen may be obliterated |
|
|
Two instances when esophagoscopy should be aborted in esophageal burn injury |
A: Cannot see the lumen A: Severe, Third Degree burn |
|
|
Five areas of extrinsic esophageal compression likely to be burned in Caustic ingestion |
A: UES – Cricopharyngeus A: Aortic arch A: Left mainstem bronchus A: LES A: Gastroesophageal junction |
|
|
Initial management strategy for Caustic Ingestions |
A: ABCs, ABG, NPO, IVF A: Judicious dilution with water or milk up to 15 ml/kg (do not induce vomiting or give neutralizing agents) A: CXR, Abdominal series A: Esophagoscopy – Timing controversial, between 24-48 hours; insertion of NG tube at time of esophagoscopy; identifies amount of damage, circumferential or not A: Gastrograffin swallow – Initial evaluation if >48 hours postingestion, to identify perforation, repeat at 6 weeks to identify stricture formation A: Steroids most useful for grade 2 injuries (1-2 mg/Kg/day, max 60 mg/day, for 21 days) A: Antibiotics controversial usage empirically (14 days) A: Antireflux medications A: Lathyrogens – Reduce collagen cross binding, to decrease esophageal stricture, unproven benefit (penicillamine, ßaminoproprionitrile, acetylcysteine) A: Sucralfate – Protects esophageal mucosa against gastric acid, and reduces granulation 3: Presence of oral injury cannot accurately predict presence or absence of more distal involvement |
|
|
Definitive management for grade 3/4 Caustic Injuries |
A: Stenting with NGT if risk of stricture (serial esophageal dilatation for the treatment of strictures) A: Thoracotomy for mural exam if difficult to determine transmural injury A: Early esophagectomy (blunt vs. thoracotomy) with reconstruction A: Esophagectomy/gastrectomy with exploratory laparotomy to remove necrotic tissue |
|
|
10 complications of caustic ingestion |
A: Stricture formation (most common) A: Tracheoesophageal fistula A: Pneumonia A: Esophageal perforation A: Mediastinitis A: Gastric perforation A: Peritonitis A: Sepsis A: Death A: Schatzki’s ring (late) A: Complete stenosis (late) A: Barrett’s esophagus (late) A: Esophageal carcinoma (late) |
|
|
Discuss Acute Laryngotracheobronchitis (Croup) |
A: Definition – Most common cause of stridor in children A: Ddx – URTI, supraglottitis, bacterial tracheitis, retropharyngeal abscess A: Diagnosis – Clinical and radiographic A: Causes – Parainfluenza type 1 (most common), 2 & 3, Influenza A, RSV, Rhinovirus, Measles, Adenovirus A: Clinical – URI prodrome, slow onset, affects 6 months – 3 years, variable/minimal fever, hoarse with barking cough, can develop respiratory difficulty with inspiratory stridor, better in supine position A: Complications – Obstruction, pulmonary edema, pneumonia, bacterial tracheitis A: Tests – Croup series, “steeple sign” on AP views A: Treatment – Expectant, humidification, racemic or levoepinephrine (0.5 ml of 2.25% solution in 3cc NS), steroids controversial (decadron 0.6-1 mg/Kg), intubation if medical therapy fails, use ETT 0.5 mm smaller than estimated, extubate when air leak detected |
|
|
Discuss Bacterial tracheitis |
A: Definition – Complication of laryngotracheobronchitis A: Ddx – of URTI, croup, supraglottits, retropharyngeal abscess A: Diagnosis – Clinical +/- bronchoscopic A: Causes – S. aureus, also S. pyogenes, H. influenzae, M. catarrhalis A: Clinical – URI prodrome, rapid onset in children 6 months – 8 years; high fever, hoarseness with cough, dysphagia, toxic symptoms, no drooling A: Tests – CBC, ABG, croup series, irregularity of airway on CXR A: Treatment – OR intubation, bronchoscopic suction and cultures of airway exudates, extubation when normothermic, decreased secretions, air leak present |
|
|
Discuss Supraglottitis (Epiglottitis) |
A: Definition – Acute inflammation of the supraglottis A: Ddx – URTI, croup, bacterial tracheitis, retropharyngeal abscess A: Diagnosis – History, clinical presentation; radiographics only if diagnosis is in question A: Causes – HiB (rare now), #1 Strept pneumo, GABHS, Staph, Klebsiella, H. parainfluenzae A: Clinical – Mild URI prodrome, rapid onset of high fever, toxic symptoms, drooling, dysphagia; affects children from 1 year to adulthood, peak is 2-6 years A: Complications – Airway obstruction, death A: Tests – Lateral soft tissue neck x-ray, “thumbprint sign”, can culture epiglottis once airway is secure A: Treatment – Do not agitate child, OR intubation, rigid/flexible bronchoscopy, IV antibiotics (ceftriaxone, cefotaxime, ampicillin/sulbactam), extubation usually within 48 hours once swelling down and air leak present |
|
|
Name all the important Nodes in the head and neck |
A: Node of Star (Submandibular) A: Rouvier (retropharyngeal) A: Delphian (Pre-tracheal) A: Virchow (Supraclav) |
|
|
Discuss Retropharyngeal abscess |
A: Definition – Purulent collection originating from the necrotic degeneration of a retropharyngeal node of Rouvier A: Ddx – URTI, croup, bacterial tracheitis, supraglottitis, parapharyngeal abscess, PTA A: Diagnosis – Clinical & radiographic A: Causes – Mixed bacteriae (Streptococci, S. aureus, H. influenzae, Bacteroides, Peptostreptococci, Fusobacteria) A: Clinical – URI prodrome in children usually <6 years, fever, sore throat, progressive dysphagia, drooling A: Complications – Airway compromise, mediastinitis, rupture & aspiration pneumonia, sepsis, arterial rupture, arterial septic emboli, jugular vein thrombosis, vertebral osteomyelitis, death A: Tests – Lateral soft tissue neck x-ray = Subcutaneous gas, widening of prevertebral tissues (>2x diameter of C2 body 90% sensitive, or >6 mm at C2 and/or >2 cm at C6, >7 mm peds/adults at C2, or >14 mm peds and >22 mm adults C6), CT scan A: Treatment – Secure airway, IV antibiotics, possible OR drainage (transoral vs. transcervical) |
|
|
Pediatric otologic development |
A: Pinna – Near adult size at 4-5 years, full size by 9 years A: Tympanic membrane – Adult sized at birth, horizontal because of incomplete ossification of EAC, vertical position reached by 2 years A: External auditory canal – Incomplete ossification at birth leads to increased compliance on impedance audiometry until age? A: Eustachian tube – 50% adult length at birth, 10 degrees, enters nasopharynx @ hard palate level; by 5-7 years lateral portion rises, tube lengthens & widens, 45 degrees, enters nasopharynx at inferior turbinate level A: Ossicles & petrous temporal bone – Adult sized at birth A: Mastoid antrum – Present at birth, increases in size during 1st year, pneumatization continues into childhood, fully developed mastoid & styloid by 3 years |
|
|
Relevant eustachian tube anatomy |
A: Length = ~36 mm in adults, ~18 mm long in infants A: 1/3 bony, 2/3 cartilaginous A: Wide at both ends, narrow in midportion at the isthmus (1 x 2 mm) A: Lateral end 4 mm above floor of epitympanum, relatively horizontal, meets cartilaginous portion at a 160 degree angle, cartilaginous portion then descends at a 40-45 degree angle, 45 degrees off sagittal, into the nasopharynx A: Torus Tubarius – Formed by soft tissue overlying the medial cartilaginous end of the tube A: Rosenmuller’s fossa – Nasopharyngeal mucosal fold found posterior to torus |
|
|
Four muscles related to the eustachian tube |
A: Tensor veli palatini/dilator tubae A: Levator veli palatini A: Salpingopharyngeus (originates off torus tubarius, interdigitates with Palatopharyngeus) A: Tensor tympani |
|
|
Tensor veli palatini anatomy |
A: Lateral head origin from scaphoid fossa & greater sphenoid wing, swings anterior, lateral & inferior, tendon around hamulus, inserts onto posterior hard palate and palatine aponeurosis |
|
|
3 functions of the Eustachian tube |
A: Pressure regulation/ventilation A: Protection from nasopharyngeal reflux A: Drainage of middle ear secretion |
|
|
JCIH Position Statement 1994 for neonatal hearing screening (FAT MOH BA) |
A: Family history of hereditary childhood SNHL A: Craniofacial Abnormalities and/or external ear malformations A: Findings associated with syndrome involving HL A: TORCHS infections A: Bacterial Meningitis A: Ototoxic medications A: Hyperbilirubinemia requiring exchange transfusion A: Birth weight under 1500g A: APGAR 0-4 at 1 minute or 0-6 at 5 minutes (Anoxia/Admission to ICU/Afterward…) A: Mechanical ventilation >5 days |
|
|
JCIH Position Statement 1994 for infant (29d-2y) hearing screening (SPIT MORE) |
A: Stigmata or other findings associated with a syndrome known to include SNHL A: Parent/caregiver concern regarding hearing, speech, language, or developmental delay A: Infections associated with SNHL A: Head Trauma with associated LOC or skull fracture A: Bacterial Meningitis A: Ototoxic medications A: Recurrent or persistent OME for at least 3 months |
|
|
The new JCIH 2007 Position Statement was released in early October 2007.18 The following is a list of some of the significant changes from the previous statement: |
A: The definition of targeted hearing loss was expanded to include neural hearing loss (eg. auditory neuropathy/dyssynchrony). A: Separate protocols are recommended for neonatal intensive care units (NICUs) and well-baby nurseries. Auditory brainstem response screenings are recommended for all NICU babies, as well as babies admitted for greater than 5 days, so that neural hearing loss will not be missed. A: Referrals should be made directly to an audiologist for comprehensive testing to include diagnostic auditory brainstem response (ABR) for all infants who do not pass ABR screening in the NICU. A: Rescreening of all infants should include re-evaluation of both ears, even if the infant only failed one ear in the initial screening. A: Audiologists with expertise in evaluating newborns should conduct diagnostic evaluations. A: Children identified with hearing loss should be fit with amplification within 1 month of diagnosis. A: A genetics consultation should be offered to families of infants diagnosed with hearing loss. A: All children identified with hearing loss should undergo an evaluation by an otolaryngologist and have at least one examination to assess visual acuity with a pediatric ophthalmologist. A: All children with any degree of bilateral or unilateral hearing loss should be considered eligible for early intervention services. A: Families should be made aware of all communication options and available hearing technologies (presented in an unbiased manner). A: Early intervention services should be provided by professionals with expertise in hearing loss. |
|
|
OAE’s for hearing screens |
A: Sensitivity ~95%, specificity ~85% A: TEOAE used most often, DPOAE being used more A: Estimates hearing in 1-6 kHz range, can go higher? |
|
|
Five Factors influencing success of OAE’s for hearing screens |
A: Noise level in test environment A: Vernix/debris in the EAC A: EAC collapse A: Middle ear fluid/mesenchyme/dysfunction A: Decreased responses in low birth weight infants & preemies |
|
|
Two Factors affecting impedance tympanography in neonates |
A: Incomplete ossification of the EAC causing greater compliance A: Persistence of Middle ear fluid or Mesenchyme |
|
|
ABR for hearing screens |
A: Sensitivity 98%, specificity 96% A: Click stimuli from 30-40 dB nHL A: Estimates hearing in 1-4 kHz range A: Requires natural sleep or conscious sedation A: Newborn ABR composed of waves I, III, V A: ABR reaches adult levels by 18 months to 3 years |
|
|
3 types of behavioral auditory testing |
A: Behavioral observation audiometry (BOA) A: Visual reinforcement audiometry (VRA) A: Conditioned play audiometry (CPA) 3: For infants/children from 0 to 5 years in age |
|
|
Behavioral observation audiometry (BOA) |
A: For infants from 0 to 6 months A: Based on reflex responses & state changes A: Unconditioned responses, variable & imprecise, habituate rapidly |
|
|
Visual reinforcement audiometry (VRA) |
A: For toddlers 6 months – 2 years A: Operant conditioning – Look for auditory stimuli (lights, motion) |
|
|
Conditioned play audiometry (CPA) |
A: For children 2-5 years A: Child performs a task in response to an auditory stimulus A: Play activity changes to keep the child’s interest |
|
|
Indications for bone conducting hearing aids |
A: Atresia/microtia A: Abnormally small EACs A: Chronic otorrhea A: Single sided deafness A: large meatoplasty/CWD mastoids that cannot fit a HA 3: For situations where air conducting hearing aids do not fulfill the amplification needs for CHL |
|
|
Four indications for imaging in pediatric hearing loss |
A: Evaluation for cochlear implantation A: Recurrent meningitis A: Sudden or progressive SNHL (especially with head trauma) A: Impact on counseling (atresia?) |
|
|
Describe the classification for congenital inner ear malformations |
A: Membranous inner ear malformations (~80%) A: Bony & Membranous malformations (~20%) A: Labyrinthine anomalies (40% of radiologically abnormal Cochleas will have SCC abnormality) A: Aqueductal anomalies A: IAC abnormalities |
|
|
Describe the pathology for Membranous inner ear malformations (3) |
A: Bing-Siebenmann – Complete membranous labyrinthine dysplasia A: Scheibe – Most common histopathologic aplasia, limited membranous cochleosaccular dysplasia (pars inferioris) A: Alexander – Partial aplasia of cochlear duct at basal turn; patients have high frequency HL |
|
|
Describe the pathology for Bony & Membranous malformations (3) |
A: Michel aplasia – Complete labyrinthine aplasia (arrest at end of 3rd week prior to Otic Capsule formation) A: Cochlear anomalies – Common cavity(Cock deformity) (4th week), Cochlear Aplasia (5th week), Cochlear Hypoplasia (6th week), A: Mondini –Incomplete bony and membranous labyrinth, cochlea may be present as a curved tube (7th week) |
|
|
What is Michel Aplasia |
A: Complete labyrinthine aplasia (3rd week arrest), rare, no hearing |
|
|
What is Common Cavity? |
A: cochlea and vestibule are confluent without internal architecture, 20%, 4th week or (abberant later week) |
|
|
What is Cochlear Aplasia? |
A: absent cochlea, rare, zero hearing, arrest in cochlear bud Formation A: still possible to implant these kids into the vestibule if an audiological response is reliably demonstrated before surgery |
|
|
Cochlear Hypoplasia? |
A: 6th week arrest, cochlea has single turn or less, 15% of cochlear deformities, variable hearing depending on membranous involvement. |
|
|
Mondini? |
A: 7th week arrest, 1.5 basal turns. Most common (50%), lack of interscalar septum, variable hearing (normal to profound deafness), organ of corti development and neural development is variable. |
|
|
Order cochlear disorders, most to least common |
A: Mondini (~50-55%) A: Common Cavity (20-25%) A: Cochlear Hypoplasia (15%) A: Cochlear aplasia (3%) A: Michel’s aplasia (1%) |
|
|
Describe the pathology for labyrinthine anomalies |
A: 40% of radiologically abnormal (osseous) Cochleas will have SCC abnormality A: SCC Dysplasia 4x as common as SCC Aplasia A: Lateral canal affected most often = Last to develop |
|
|
Describe the pathology for Aqueductal anomalies |
A: VA normally 0.4-1 mm in diameter when measured halfway between common crus and external aperture A: Enlarged VA >1.5 mm, usually bilateral, typically born with normal to mild SNHL and ~40% eventually develop Profound SNHL; prone to sudden SNHL with head trauma A: Enlarged CA to be diagnosed radiographically, the intraosseous portion coursing toward the vestibule must be enlarged beyond 1 mm; external aperture measures 3-4 mm and even beyond 6 mm |
|
|
Describe the pathology for IAC abnormalities |
A: Narrow IAC <3 mm, if facial function present then CN VIII likely Absent A: Widened IAC >10 mm, associated with stapes Gusher |
|
|
Mechanism of hearing loss in EVAS? |
A: Hydrostatic Forces are transmitted either 1) from the endolymphatic sac through an enlarged endolymphatic duct or 2) from the subarachnoid space through cochlear modiolar defects to hair cells within the cochlea damaging them A: 2nd theory is that the endolymphatic sac contains fluid with abnormal osmolarity and that reflux of this fluid into cochlea by way of a patent endolymphatic duct damages the neuroepithelium |
|
|
lassification of Microtia |
A: Class I – Mild deformity, auricle decreased in size A: Class II – Curving vertical ridge of tissue, all major structures present but with absolute deficiency of tissue A: Class III – Rudimentary soft tissue structure, no recognizable auricle or canal (peanut ear) A: Class IV - anotia |
|
|
Classification of EAC/ME Deformity (Ombredanne) |
A: Minor – EAC may be mildly stenotic, ME space & TM normal or slightly small, conductive hearing loss secondary to ossicle absence or fixation A: Major – EAC & TM usually absent, decreased or absent middle ear space, ossicles rudimentary or absent, fused or fixed to atretic plate, CN VII displaced |
|
|
Schuknecht classification of aural atresia (1989)? |
|
|
|
Altmann’s classification of atresia (1955) |

|
|
|
Four criteria important in the De la Cruz classification of congenital aural atresia
|
A: Mastoid pneumatization A: Oval window footplate A: Footplate/CN VII relationship A: Inner ear 3: Normal in Minor, abnormal in Major |
|
|
Describe Jahrsdoerfer’s grading system for congenital aural atresia |
A: Presence of the stapes (2 points) A: Oval window status A: Round window status A: Facial nerve course through the middle ear A: Malleus/incus complex A: Incus-stapes connection A: Middle ear space A: Mastoid pneumatization A: External ear appearance 3: 10 point system based on high resolution CT findings of the temporal bone; ≤5 = not surgical candidates, ≥8 = 80% success |
|
|
Two audiometric tests useful for bilateral aural atresia |
A: Bone conduction ABR – Wave I generated at distal CN VIII, little crossover A: ECOG – For minor atresia, with transtympanic, TM, or EAC electrode |
|
|
Six Syndromes associated with Microtia/Aural Atresia |
A: CHARGE A: Goldenhar’s/Hemifacial macrosomia A: Treacher-Collins A: Crouzon’s disease A: Pierre-Robin sequence A: Branchio-oto-renal |
|
|
General management approach for Aural Atresia |
A: BC ABR/ECoG – For Unilateral to confirm other ear is normal, and for Bilateral to evaluate cochlear function A: High resolution CT of the temporal bone at age 5-6 A: Uniateral microtia & aural atresia – Do not reconstruct canal, as speech & learning will develop normally A: Bilateral aural atresia – Begin with the Better developed ear as child approaches school age, and depending on the results, may proceed with second ear within next few years A: Defer surgery to 6-8 years, allows growth of contralateral ear (template) and rib cage (cartilage graft source) A: Bone conduction hearing aids for Bilateral cases |
|
|
Four criteria for surgery in Aural Atresia |
A: Presence of Cholesteatoma – Emergent indication, and reason every aural atresia requires CT at some point A: Normal Sensorineural function (just CHL) A: Ossicular mass present A: Middle ear space at least 50% normal size |
|
|
Two absolute and 1 relative contraindications to surgical correction of Aural Atresia |
A: Abnormal inner ear Morphology demonstrated on CT scan A: Abnormal Cochlear function demonstrated by audiologic testing A: Jahrsdoerfer score ≤5/10 (relative) |
|
|
Two approaches to Aural Atresia repair |
A: Transmastoid approach A: Anterior approach |
|
|
Four facial nerve findings in middle ear Atresia |
A: Tympanic segment dehiscence A: Inferior/Lateral displacement of the tympanic segment A: Anterior/Lateral displacement of the mastoid segment A: Therefore, more acute angle taken at 2nd genu (60 degrees vs. 90-120 normally) |
|
|
Five complications in Atresia surgery |
A: EAC stenosis A: Recurrent/persistent CHL A: SNHL A: Facial nerve injury A: Chronic infection 3: Risks minized by near normal CN VII course, middle ear and mastoid at least 2/3 normal size |
|
|
Audiometric results of Aural atresia repair |
A: HL <30 dB in 50-75% A: HL <20 dB in 15-50% |
|
|
Classification of Otitis |
A: Acute otitis media (AOM) – 1) Acute Onset of signs & symptoms, 2) Presence of MEE, 3) Signs & symptoms of middle ear Inflammation (include bulging or fullness of the tympanic membrane (TM), erythema of the TM, and acute perforation of the TM with otorrhea) (Symptoms include otalgia, irritability, and fever) A: Recurrent Acute otitis media (R-AOM) – ≥3x/6 mo or ≥4x/1 yr; recurrences in <1 mo = Same pathogen, in >1 mo = Different strain A: Otitis media with effusion (OME) – MEE without signs and symptoms of acute inflammation as found in AOM. A: Chronic Suppurative otitis media: CSOM is defined as chronic otorrhea (ie, lasting >6-12 wk) through a perforated tympanic membrane |
|
|
Ten host risk factors for AOM |
A: Age (<2 yo, first onset <6 mo) A: Gender (Male) A: Race (First Nations) A: Genetic predisposition A: Ciliary dyskinesia A: Adenoids (reservoir of infection & mechanical ET obstruction) A: ET dysfunction (short, horizontal, compliant) A: Cleft palate, Craniofacial abnormality, Down’s A: Immune deficiency A: Atopy (disputed) A: +ve family Hx |
|
|
Ten Environmental risk factors for AOM |
A: Daycare attendance (2.6x) A: Season (Fall/Winter) A: URTIs A: Older siblings A: Parental history of OM A: Passive smoking A: Low S/E status (overcrowding, poor sanitation) A: Lack of breastfeeding A: Night-time bottle (horizontal position) A: Pacifier use |
|
|
Virology of AOM (4 – RRIP!) |
A: RSV (#1) - paramyxovirus A: Rhinovirus A: Influenza A: Parainfluenza |
|
|
Bacteriology of AOM |
A: Streptococcus pneumonia A: Heamophilus influenza A: Moraxella catarrhalis A: Streptococcus pyogenes 3: Far less common are S. Aureus and GNBs |
|
|
Ten Intratemporal complications of AOM |
A: Chronic suppurative OM A: Adhesive otitis A: Tympanic membrane perforation A: Cholesteatoma A: Tympanosclerosis A: Fixation and Discontinuity of ossicular chain A: Mastoiditis with or without Abscess (Postauricular, Bezold’s, Zygomatic, Parapharyngeal, Retropharyngeal) A: Petrositis A: Labyrinthitis – Serous or Suppurative A: Facial palsy A: Labyrinthine fistula (Pasha) A: CHL or SNHL |
|
|
Bacteriology of Acute Mastoiditis not due to cholesteatoma |
A: Streptococcus pneumonia A: Heamophilus influenza A: Streptococcus pyogenes A: Coag negative Staph A: Staphylococcus aureus (#3?) 3: Pseudomonas #2 in Bailey (in cholesteatoma?) |
|
|
Bacteriology Chronic Otorrhea with or without cholesteatoma |
A: Mixed flora including Streptococcus pneumonia and Anaerobes (including Bacteroides) A: Pseudomonas (most common aerobe) A: Staphylococcus aureus and epidermidis A: Gram negatives – Klebsiella, E. coli, Proteus |
|
|
6 Intracranial complications of AOM |
A: Meningitis (#1, HiB > Pneumococcus, Mondini) A: Epidural/subdural/cerebral abscesses A: Focal encephalitis A: Lateral/sigmoid sinus thrombosis (Tobey-Ayer/Quesckenstedt’s Test; Griesinger’s sign)) A: Otitic hydrocephalus A: Blindness with optic neuropathy |
|
|
Bacteriology of bacterial Meningitis |
A: Heamophilus influenza (non-typable) A: Streptococcus pneumonia A: Nisseria meningitidis |
|
|
Bacteriology of intracranial Abscess |
A: Streptococcus sp. (pneumonia, pyogenes) A: Staphylococcus sp. A: Proteus sp. A: Anaerobes (Peptococcus, Peptostreptococcus, B. fragilis) |
|
|
Five early Signs of Impending Complication |
A: Persistence of acute infection for 2 weeks A: Recurrence of symptoms within 2 weeks A: Fetid discharge during treatment A: Acute exacerbation of chronic infection, especially if fetid A: HiB or Anaerobes 3: “Two Week Fetid Exacerbation with Type B Anaerobes” |
|
|
Criteria for 72 hour observation for AOM |
A: Appropriate F/U A: Otherwise healthy children (see notes) A: Abx started if SSx persist/worsen 3: Healthy = no immune deficiency/craniofacial anomalies, no treatment failures/relapse within 30 days, no co-existing acute sinusitis/streptococcal pharyngitis, no underlying chronic OME, no complicated AOM |
|
|
Pathogen causing bilateral, dull TM, non-mobile, T > 37°C |
A: Heamophilus influenzae |
|
|
Pathogen causing unilateral, bulging TM, T >100.6 (38.1)° |
A: Streptococcus pneumoniae |
|
|
Pathogen causing bullous myringitis |
A: Streptococcus pneumoniae 3: Others include Mycoplasma, H. flu, Beta-hemolytic strep, M. catarrhalis, Parainfluenza & influenza virus??? |
|
|
Pathogen causing spontaneous perforation of ™ |
A: Streptococcus pyogenes |
|
|
Eight Indications for PETs |
A: Recurrent AOM (>3/6 mo, >4/1 yr, with failed medical management) A: OME (bilateral >3 mo or unilateral >6 mo; earlier when >25db CHL, speech/language delay, severe retraction pocket, disequilibrium/vertigo, or tinnitus present) A: Eustachian tube dysfunction with autophony, disequilibrium or vertigo, tinnitus, or Atelectatic TM/severe retraction pocket, unrelieved by medical management A: Patulous eustachian tube A: Barotitis media/Hyperbaric oxygen therapy A: Suspected unusual pathogen A: Suppurative complication, present or suspected A: Unsatisfactory response to antibiotics |
|
|
Five Indications for Tympanocentesis/Myringotomy |
A: AOM in a seriously-ill, toxic, newborn or immune deficient patient A: Severe pain A: Suppurative complications (facial paralysis, meningitis, etc.) A: Unsatisfactory response to antibiotics A: Suspected unusual pathogen (newborns, immunodeficiency) |
|
|
Nine complications of Tympanostomy tubes |
A: Otorrhea: ~3.5% rate of persistent drainage A: Granulation tissue formation A: Myringosclerosis A: TM perforation A: TM atrophy, retraction, atelectasis A: Cholesteatoma A: Loss of tube in the middle ear A: Early extrusion A: Plugged tube |
|
|
Etiologies of pediatric hearing loss and percentage of cases? |
A: Genetic = 50% (30% part of an identifiable Syndrome, AR ~75%, AD ~20%, XL ~3-4%, Mitochondrial <1%) A: Environmental = 20-25% (pre-, peri-, postnatal) A: Unknown etiology = 25-30% |
|
|
Percentage of Genetic hearing loss that is Syndromic |
A: ~30% |
|
|
Discuss connexin 26 (CX26) |
A: Gene GJB2 product A: DFNB1 mutation Most common association with Genetic Nonsyndromic hearing loss (30% of Sporadic HL, >50% of cases with an affected Sibling) A: Six connexins form a connexon |
|
|
Mondini malformation is associated with which 6 syndromes? |
A: Mobius syndrome A: Branchiootorenal A: Waardenburg A: Pendred A: Treacher-Collins A: Klippel Feil (Wildervanck) 3: Wild Wars Teach Brothers Patience |
|
|
Scheibe malformation is associated with which 4 syndromes? |
A: Jervell-Lange Nielsen A: Refsum (RP, ataxia, SNHL) A: Usher A: Waardenburg 3: “JR Ushers Warden” |
|
|
Name some Autosomal Dominant syndromes associated with SNHL |
A: Branchiootorenal syndrome (Melnick-Fraser) A: Stickler syndrome A: Waardenburg syndrome A: Osteogenesis imperfecta (van der Hoeve syndrome) A: Otosclerosis A: Neurofibromatosis A: Treacher-Collins syndrome 3: WB TOONS Dominate |
|
|
Describe Waardenburg’s syndrome and the 4 types |
A: Definition – Autosomal dominant except type IV = AR A: Clinical – Pigment abnormalities (white forelock, premature graying, vitiligo, heterochromia iridis), craniofacial abnormalities (dystopia canthorum, broad nasal root, synophrys), unilateral or bilateral SNHL, +/- vestibular Sxs A: Type I – Presence of Dystopia Canthorum, SNHL occurs in 20%, PAX3 gene on chromosome 2 Type II – Absence of dystopia canthorum, SNHL occurs in 50%, MITF (microphthalmia transcription factor) on chromosome 3 Type III – Klein-Waardenburg, features of WS1 plus Blue eyes, Hearing impairment, upper limb Skeletal dysplasias, muscular hypotonia, unilateral ptosis? (Pasha) Type IV – Waardenburg-Shah, AR, phenotype similar to WS2 plus Hirschprung megacolon |
|
|
Describe Neurofibromatosis Type I (von Recklinghausen’s disease) |
A: Mutation of nerve growth factor Neurofibromin on chromosome 17 A: CNS involvement may lead to MR, blindness, SNHL, acoustic neuromas present in only 5% |
|
|
Diagnosis of NF Type I by 2 of the following 7 criteria |
A: Neurofibromas (2 or more cutaneous, or 1 plexiform) A: Café au lait spots (6 or more, >15 mm in adults, >5-10 mm in children) A: Axillary or inguinal freckling A: Lisch nodules (2 or more iris hamartomas) A: Optic glioma A: A distinctive osseous lesion– pseudoarthrosis, scoliosis, or thinning of the tibia; sphenoid wing dysplasia A: Family history or 1st degree relative with confirmed NF1 |
|
|
Describe Neufibromatosis type II |
A: Mutation of tumor suppressor gene Merlin on chromosome 22q A: Autosomal dominant, but 50% of cases due to spontaneous mutations A: 95% incidence of Bilateral Acoustic Neuromas often before age 21 A: Central Meningiomas, Gliomas, Schwannomas, early lens opacifications (cataracts) A: Fewer café au lait spots & cutaneous nodules than NF I |
|
|
Neufibromatosis type II subtypes (2) |
A: Wishart – Early onset, rapid growth, other tumors than acoustic neuromas A: Gardner – Slower rate of growth & onset, usually only bilateral acoustic neuromas |
|
|
Diagnosis of NF Type II by 1 of the following 3 criteria |
A: Bilateral CN VIII masses on MRI (seen with & without Gado, on Axial and Coronal cuts) A: Family history or 1st degree relative with confirmed NF2, and 1 CN VIII mass (as described above) A: Family history or 1st degree relative with confirmed NF2, and at least 2 of 5: Neurofibroma, Meningioma, Schwannoma, Glioma, Juvenile Cataracts (Posterior Sub-Capsular) |
|
|
Name 3 autosomal recessive syndromes associated with SNHL (PUJ) |
A: Pendred’s syndrome A: Usher syndrome A: Jervell-Lange Nielsen syndrome 3: PUJ goes to recess |
|
|
Describe Usher syndrome and the 4 types |
A: Definition – Most common cause of congenital visual deafness; Autosomal recessive except type IV = XR, 10% of hereditary deafness A: Diagnosis – Eye changes detected on Electroretinography even before funduscopic changes A: Clinical – Retinitis pigmentosa (progressive visual loss), atrophy of organ of Corti/Scheibe aplasia (congenital deafness), Ataxia and vestibular dysfunction common A: Complications – Deafness, blindness A: Type I – Profound congenital deafness, RP onset by age 10, no vestibular response, 90% of cases Type II – Moderate/severe congenital deafness, onset of RP in teens/twenties, normal or decreased vestibular response, 10% of cases Type III – Progressive HL, RP begins in puberty, <1% of cases Type IV – X-linked inheritance, similar to type II A: Tests – Electroretinography (measures potentials of retinas from light & visual stimuli), Opthalmology consult essential A: Treatment – Amplification |
|
|
Describe Jervell Lange-Nielsen disease |
A: Definition – Autosomal recessive A: Causes – Linked to a Potassium channel gene (KVLQT 1) on 11p15.5 A: Clinical – Profound bilateral congenital SNHL (high frequencies worse), Heart disease (prolonged QT, large T waves) A: Complications – Stokes-Adams attacks, recurrent syncope, usually terminates fatally with sudden death A: Tests – ECG, Audiometry A: Treatment – ß-blockade, Amplification |
|
|
Describe Pendred syndrome |

A: Definition – Autosomal recessive A: Causes – Tyrosine Iodination defect leading to Euthyroid goiter A: Clinical – Goiter, Mondini or Enlarged VA’s A: Tests – Perchlorate discharge test A: Treatment – Exogenous T4 |
|
|
Name 4 X-linked Recessive syndromes associated with SNHL
|
A: Wildervaank syndrome A: Otopalatodigital syndrome A: Alport syndrome A: Norrie syndrome A: Deafness Dystonic 3: WANDO loves SEX |
|
|
Describe Wildervank syndrome |
A: Definition – XR, Klippel-Feil malformation, Brevicollis, mode of inheritance unclear A: Clinical – SNHL or mixed loss, due to bony inner ear (Mondini) and middle ear malformation, CN VI paralysis with eye retraction on lateral gaze (Duane retraction syndrome), short neck/fused cervical vertebrae, Assimilation of the Atlas (Basilar impression), Spina bifida |
|
|
Discuss Otopalatodigital syndrome |
A: Definition – XR A: Causes – Mutation localized to Xq28 A: Clinical – CHL due to ossicular malformation, Cleft palate, Stubby/clubbed digits, Craniofacial deformities (hypertelorism, supraorbital deformity, flat midface, small nose), short stature, wide space between 1st & 2nd toe |
|
|
Discuss Alport syndrome |
A: Definition – AR or XR, abnormal Type IV collagen in the glomerular basement membrane A: Diagnosis – Renal failure and SNHL A: Causes – Degeneration of organ of Corti and Stria vascularis A: Clinical – Progressive SNHL (usually 2nd decade), varying degrees of Renal disease (mild renal Dysplasia to renal Agenesis identifiable on ultrasound), gross Hematuria associated with UTI A: Complications – Renal failure, Death in males by age 30 A: Types – 6 subtypes, 3 AR, 3 X-linked A: Tests – BUN, Creatinine, Urinalysis A: Treatment – Dialysis/renal transplant important therapies (more so in males) |
|
|
Discuss Norrie syndrome |
A: Definition – XR A: Causes – Mutation of NDP gene on Xp11.4, produces norrin protein, structurally similar to TGF-ß A: Clinical – Progressive SNHL (2nd/3rd decade), occasional progressive mental deterioration, congenital/rapidly progressive Blindness due to pseudoglioma, exudative vitreoretinopathy, opacification, ocular degeneration |
|
|
11 lab tests and there association with childhood SNHL |
A: CBC + Diff – Leukemia/lymphoma; Platelets – Fechner syndrome (Very rare, HF SNHL, Ocular disease, Proteinuria, Macrothrombocytopenia) A: BUN, CR, Urinanalysis – Alports syndrome (persistent microscopic hematuria) A: Glucose – Alstron syndrome (Obesity, Impaired glucose tolerance with insulin resistance, Retinal degeneration, Neurosensory deafness, Acanthosis nigricans, Hepatic dysfunction, and other Endocrine abnormalities) A: RPR, VDRL,FTA-ABS – Syphilis A: EKG – Jervell and Lange-Nielson syndrome A: GJB2 gene – Responsible for 50% of autosomal recessive nonsyndromic hearing loss, found to be positive in 35% of moderate to profound SNHL. A: CT scan – Michel aplasia, Mondini malformation, Enlarged VA A: MRI – Child with NF II, useful if progressive hearing loss, or vestibular symptoms, focal neurological symptoms A: ANA, ESR, RF – SLE, RA A: Thryoid function tests – Cretinism and Pendred syndrome (Pendred may have normal thyroid function tests) |
|
|
Which 4 tests should be routine when no other etiology of childhood SNHL is found? |
A: EKG A: RPR A: CT A: GJB2 3: In addition to CBC, SMA7 (may also detect leukemia, Fechner, Alstron, and Alport) |
|
|
Most common cause of chronic benign pediatric lymphadenopathy, diagnosis, and treatment |
A: Cat Scratch Disease (Bartonella henselae) A: Serologic testing A: Intracellular gram-negative bacillus on Warthin-Starry stain A: Azithromycin (or self limiting) 3: Avoid I+D (draining tract) |
|
|
Condition in immunocompromised patients with B. henselae infection |
A: Bacillary angiomatosis |
|
|
Typical presentation of atypical mycobacterium lymphadenitis, most common organism, and treatment |
A: Submandibular lymphadenopathy, most common in under 12, draining sinus if untreated, later calcifies A: M. avium intracellulare A: Surgical excision, can try Antibiotic triple therapy (Clarithromycin, Rifampin, & Ethambutol) if disseminated disease |
|
|
Electron microscopy finding of Histiocytosis X (LCH) |
A: Bierbeck granules 3: “Tennis-racket” or rod shaped cytoplasmic organelles with a central linear density and a striated appearance |
|
|
Define Epipericardial ridge, 4 muscles and 2 nerves which are derived from it |
A: An elevation of mesoderm separating the developing pharyngeal region from the embryonic pericardium A: Forms SCM, Trapezius, Tongue and Infrahyoid musculature A: Nerves present include XI and XII |
|
|
Proper surgical management of Type II First Branchial arch abnormality |
A: Superficial parotidectomy A: CN VII identification A: Trace track back to origin (typically the bony/cartilaginous junction of EAC) A: Cartilage resection 3: Relation to CN VII variable |
|
|
3 and 4th Branchial abnormalities can manifest as what condition in children |
A: Acute suppurative thyroiditis |
|
|
What is CHAOS and two example of lesions that can cause conditions |
A: Congenital High Airway Obstruction Syndrome A: Lymphangiomas and Teratomas located in the anterior compartment |
|
|
CHAOS – how diagnosed and findings |
A: Ultrasound findings in utero for diagnosis A: Dilated airways below the level of the upper airway obstruction, large echogenic lungs, flattened diaphragms, fetal hydrops, dilated tracheobronchial tree |
|
|
Define EXIT procedure |
A: Ex utero intrapartum treatment procedure for babies with airway compression, partially delivered through a C-section but remain attached by their umbilical cord to the placenta, while a pediatric or neonatal general surgeon establishes an airway so the fetus can breathe, the umbilical cord is cut and clamped, and the infant is fully delivered. Then the remainder of the C-section proceeds |
|
|
Most common infectious organism in pediatric bacterial cervical lymphadenitis |
A: Staphylococcus aureus A: Streptococcus pyogenes |
|
|
Bacterial cause of Lemierre’s Syndrome |
A: Fusobacterium necroforum |
|
|
Non-tuberculous vs tuberculous lymphadenopathy in Pediatrics |
A: Systemic symptoms – Single large node with fevers and malaise in TB; Rare in NTB A: Lung disease – Often in TB; Rare in NTB A: Violaceous skin change in NTB A: PPD – Strongly positive in TB; weak or negative in NTB A: Treatment – Medical in TB; NTB often resistant to medical, therefore often require Surgical excision |
|
|
Most Sensitive tests for Mononucleosis |
A: IgM(current and recent disease) and IgG(current and past disease) for EBV (VCA and EBNA) 3: Monospot/Heterophile Ab test is negative in early disease (for 2- 3 weeks) |
|
|
Percentage of pediatric NHL that are high grade |
A: 90% |
|
|
Bifid epiglottis – what other test required |
A: Thyroid function (dysgenesis in Bamforth syndrome) 3: Also associated with Pallister-Hall syndrome |
|
|
Classification of laryngeal (saccular) cysts |
A: Superior (extending into ventricle) A: Posterior (extending into the AE folds and false cord) |
|
|
Ddx of congenital supraglottic abnormalities (8) |
A: Laryngomalacia A: Hemangioma A: Lymphatic malformation A: Laryngocele A: Saccular cyst A: Anomalous cuneiform cartilage A: Bifid epiglottis A: Supraglottic web |
|
|
Six techniques for improving airway in bilateral vocal cord paralysis |
A: CO2 laser cordotomy A: Laser arytenoidectomy and cordectomy A: Open arytenoidectomy A: Arytenoidopexy A: Arytenoid separation with cartilage grafting A: Laryngeal reanimation techniques (phrenic to RLN, phrenic to PCA, or omohyoid muscle pedicle) |
|
|
Define Pallister-Hall syndrome |
A: Laryngeal (clefts or bifid epiglottis) A: Hypothalamus abnormalities (hypothalamic hamartoblastoma, hypopituitarism) A: Postaxial polydactyly A: Imperforate anus |
|
|
Age 1 and recurrent croup – what investigation |
A: Endoscopy – R/O subglottic stenosis |
|
|
Two conditions with congenitally shallow orbits |
A: Crouzons A: Aperts A: Pfeiffer |
|
|
Ddx of chronic drooling |
A: Medications A: Neurologic A: Indirect causes – Nasal obstruction, head position, malocclusion, tongue size, sitting position, and emotional state |
|
|
Treatment options for Drooling |
A: Speech therapy A: Behavior therapy A: Dental appliance A: Drugs (glycopyorolate, trihexyphenidyl, scopolamine, Botox) A: Radiation A: Surgery – SMG excision or Duct re-routing Parotid duct ligation, Tympanic neurectomy, Intraductal laser photocoagulation |
|
|
AAO-HNS indications for coagulation studies |
A: Only if indicated by history or if genetic information is unavailable |
|
|
Location and type of mutation associated with VCF |
A: Chromosome 22q11.2 A: Hemizygous Microdeletion |
|
|
Normal parents, with child with SNHL, chance of further children with SNHL (all comers) |
A: 14% |
|
|
Black child for T and A – what need to consider first, simple test and preoperative management |
A: Consider Sickle Cell anemia; 3 forms vaso-occlusive (painful) crises, aplastic crises, and splenic sequestration crises A: Simple test – CBC (anemia), Sickledex solubility test, then Hb electrophoresis A: Preop – Consult hematology, Evidence of renal, pulmonary or cerebrovascular complications should be assessed pre-operatively. Check for signs of vaso-occlusion, fever, infection and dehydration. transfusion to achieve a ratio of 70:30 for normal blood to HbS to help prevent Acute chest syndrome post op and avoid hypoxia and precipitate a sickle cell crisis |
|
|
Diagnosis of Macroglossia – any of 3 following criteria |
A: Extravasation of lingual apex or lingual border onto or outside the dentition A: Impression of ≥ 1 teeth on lingual border visualized when mouth is open A: Following Sx for correction, a relapse of increased interdental space, open bite deformity, and/or jaw deformation with malocclusion occurs |
|
|
Macroglossia triad |
A: Open bite deformity A: Mandibular prognathism A: Malalignment |
|
|
Classification of Local causes of Macroglossia (CITeN) |
A: Congenital – Hemangioma, Lymphangioma, Lingual thyroid A: Inflammatory/Infectious – Angioedema, TB, Actinomycosis, Dental infection, Syphilitic gumma, Riga disease, Ranula A: Traumatic – Dental irritation, Hematoma, Postoperative edema, Sublingual calculus A: Neoplastic, Benign – Granular cell tumour, Neurofibroma, Leiomyoma, Lipoma; Malignant – Carcinoma, Sarcoma |
|
|
Classification of Generalized causes of Macroglossia (CITEn) |
A: Congenital – Primary idiopathic macroglossia, Beckwith- Wiedemann syndrome, Down syndrome, Trisomy 4P syndrome, Triploid syndrome, Gangliosidosis syndrome, Mucopolysaccharidoses, Robinow syndrome A: Inflammatory – Chronic glossitis A: Toxic – Amyloidosis, Lipoid proteinosis, Chronic steroid therapy A: Endocrine – Acromegaly, Myxedema, Cretinism |
|
|
Three causes of Pseudo-Macroglossia |
A: Habitual tongue posturing A: Retrognathia/Micrognathia A: Hypotonia of tongue |
|
|
Four treatment options for macroglossia |
A: Observation A: Orofacial therapy (for hypotonia, or lingual mal-position) A: Surgery A: Submucosal minimally invasive lingual excision (SMILE) |
|
|
Five indications for Macroglossia Surgery |
A: Airway obstruction – BOT macroglossia A: Speech impediment - particularly consonants requiring tip contact w/ alveolar ridge, palate A: Dysphagia – failure to thrive A: Maxillofacial abnormalities – anterior open bite, prognathism, increased ramus-body angle A: Cosmesis |
|
|
Surgical options for Macroglossia |
A: Anterior wedge A: Tip amputation A: Dorsum and tip A: Central reduction A: Block excision central & tip or Keyhole A: Tip preservation – Kruchinsky A: Horizontal filleting A: Dorsal flap |
|
|
Two indications for surgery in Ankyloglossia |
A: Poor feeding/sucking – First few months A: Impaired speech – 2-3 years |