Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
473 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Regular visits schedule
|
First week after discharge :
- 1, 2, 4, 6, 9, 12, 15, 18 & 24 Months - Yearly until age 6 - Every other year after Yearly again after age 11 |
|
|
|
Mnemonic for paediatric developmental milestones
(1,2,3,4 years) |
1 year
- single words 2 years - 2 word sentences - Understands 2 step commands 3 years - 3 word combos - Repeats 3 digits - Rides tricycles 4 years - Draws square - Counts 4 objects |
|
|
|
Speech and language development in children
|
2 MO : coos
4 MO : Responds to voice, laugh 6 MO : Begins to babble, responds to name 9 MO : "Mama, Dada", imitates words 12 MO : 2 words, follow 1 step commands 15 MO : Jargon 18 MO : 10 words, follows single commands 24 MO : 2-3 words sentences 3 years : Prepositions, plurals, counts to 10, 75% intelligible 4 years : Tells story, knows 4 colours, speech intelligible, uses past tense |
|
|
|
Gross motor developmental milestones
|
6 MO : tripod sit
9 MO : pulls to stand 12 MO : walks with support 15 MO : walks without support 18 MO : up steps with help 24 MO : runs, kicks ball, walks up and down steps |
|
|
|
Primitive reflexes and significance
|
Reflexes seen in normal newborns. They may indicate abnormality if they persist after 4-6 months.
1) Moro reflex - infant is placed semi-upright, head supported by examiner's hand, sudden withdrawal of supported head with immediate resupport elicit reflexes. - reflex consists of abduction and extension of the arms, opening of the hands, followed by flexion and adduction of arms. - absence of Moro suggests CNS injury; asymmetry suggests focal motor lesions (e.g. brachial plexus injury). 2) Galant reflex - infant is held in ventral suspension and one side of the back is stroked along the paravertebral line; the pelvis will move in the direction of stimulated side. 3) Grasp reflex - flexion of fingers with the placement of a finger in the infant's palm 4) Tonic neck reflex - turning the head results in the "fencing" posture (extension of ipsilateral leg and arm) 5) Primitive walking : - infant places foot on a surface when it is brought into contact with it 6) Rooting reflex : - infant pursues tactile stimuli near the mouth 7) Parachute reflex : - tilting the infant to the side while in a sitting position results in ipsilateral arm extension (appears by 6-8 MO) 8) Babinski sign is normal in infants (< 2 years) |
|
|
|
Routine Québec immunization calendar
|
2 MO : DCaT-HiB-Polio, Pneu-C
4 MO : DCaT-HiB-Polio, Pneu-C 6 MO : DCaT-HiB-Polio, Influenza 1st shot 7 MO : Influenza 2nd shot 12 MO : RRO, Varicella, Men-C, Pneu-C 18 MO : RRO, DCaT-HiB-Polio 4-6 Yrs : DCaT-Polio 14-16 Yrs : DCaT, then every 10 years 4th primary year : HBV + HPV (for girls) |
|
|
|
Which vaccines should be administered at the same visit or separated by 4 weeks or more?
|
The two live vaccines :
- MMR - Varicella |
|
|
|
Schedule for DCaT-HiB-Polio immunization.
|
2,4,6,18 MO
DCaT-Polio 4-6 Yrs DCaT 14-16 Yrs, then every 10 years |
|
|
|
Schedule for Pneu-C vaccination
|
2, 4 & 12 MO
|
|
|
|
Schedule for Influeza vaccination
|
6, 7 MO
|
|
|
|
Schedule for Men-C vaccination
|
12 MO
|
|
|
|
Schedule for RRO vaccination
|
12, 18 MO
|
|
|
|
Schedule for Varicella vaccination
|
12 MO
|
|
|
|
Discuss the safety of the MMR vaccine
|
According to the CDC, the weight of currently available scientific evidence does not support the hypothesis that MMR vaccine causes either autism or IBD.
|
|
|
|
Contra-indications to any vaccine
|
Moderate to severe illness with or without fever
Allergy to vaccine component No need to delay fever for mild URTI |
|
|
|
Conduct if mother is HBsAg +
|
Give HBIg at birth and Hep B vaccine at birth, 1 MO and 6 MO
|
|
|
|
Pediatrics
Positive TB test |
> 15 mm : children > 4 years with no risk factors
> 10 mm : children < 4 years, or at risk for environmental exposure > 5 mm : children with close TB contact, immunosuppressed |
|
|
|
Indication for BCG vaccine
|
Infants of parents with infectious TB at time of delivery
Groups with high rates of disease Only given if patient has negative TB skin test Possible side effects : erythema,papule formation 3-6 weeks, enlargement of regional lymph nodes |
|
|
|
Safety and efficacy of an attenuated vaccine against severe rotavirus gastroenteritis
|
The vaccine is 85% efficacious against severe rotavirus gastroenteritis and hospitalizations associated with gastroenteritis and 100% against more severe gastroenteritis.
NEJM 2006;354:11-22 |
|
|
|
Signs of inadequate intake
|
< 6 wet diapers per day after first week
sleepy or lethargic < 7 feeds per day sleeping throughout the night weight loss > 10% of birth weight |
|
|
|
Which supplements do breast fead babies require?
|
Vitamin K (given IM at birth)
Vitamin D 400 IU; especially during winter months Fluoride (after 6 MO if not sufficient in water supply) Iron : from 4 months to 12 months |
|
|
|
Contra-indication to breast feeding
|
Mothers receiving chemotherapy or radioactive compounds
Mothers with HIV / AIDS, active untreated TB, Herpes in breast region Mothers using > 0.5 g/kg/day alcohol and / or illicit drugs Mothers taking certain medications e.g. antimetabolites, bromocriptine, chloramphenicol, high dose diazepam, ergots, gold, metronidazole, tetracycline, lithium, cyclophosphamide ... etc. N.B. Oral contraceptives are not a contraindication for breast feeding |
|
|
|
Advantages of breast feeding
|
Breast milk is easily digested and has low renal solute load.
Immunologic benefits - IgA, macrophages, active lymphocytes, lysozymes (lactoferrin inhibits E. Coli growth in intestine) - Protection is greatest during early months, but is cumulative with increased duration of breastfeeding - Lower allerginicity than cow's milk protein (decreased cow's milk protein allergy and eczema) - Lower pH promotes growth of lactobacillus in the GI tract Parent child bonding Economical Convenient Retards return of menstrual cycle |
|
|
|
Infant growth and health outcomes associated with 3 months compared with 6 months of exclusive breastfeeding.
|
There is an association between breastfeeding and a lower incidence of gastrointestinal infections in term infants.
|
|
|
|
Complications of breast feeding
|
Mother
- Sore / cracked nipples : treat with warm compresses, massage, frequent feeds, soothing barrier creams and proper latching technique - Breast engorgement (usually in first week) : continue breast feeding and or pumping - Mastitis : treat with cold compresses between feeds, cloxacillin for mother, continue nursing +/- incision and drainage. Infant - Breast feeding jaundice (first 1-2 weeks) : due to lack of milk production and subsequent dehydration - Breast milk jaundice : rare (0.5% of newborns); due to substances in breast milk that inhibit conjugation of bilirubin (persists up to 4-6 months) - Poor weight gain : consider dehydration or failure to thrive - Oral candidiasis : check baby's mouth for white cheesy material that does not scrape off; treat baby with antifungal such as nystatin (Mycostatin) + treat mother topically to prevent transmission. |
|
|
|
Indications for cow's milk
|
Premature babies
Transitional Contraindication to breastfeeding Cow's milk has plant fats instead of dietary butterfat and a lower whey:casein ratio. It also reduces the absorption |
|
|
|
Fortified formula indications and content
|
Low birth weight and premature babies
__________________________________________________________ More Calories Higher amounts of vitamins A, D, E & K May only be used in hospital due to risk of fat soluble vitamin toxicity. |
|
|
|
Nutrition of infant
|
• De 0 à 6 mois : allaitement exclusif
• De 0 à 12 mois : suppléments vitamine D 400 UI • À 4 mois, le bébé est prêt à commencer à digérer des solides. Il faut commencer à les offrir vers l’âge de 6 mois, en petite quantité de 2 à 3 fois par jour. • On commence un nouvel aliment à la fois, à une semaine d’intervalle. • L’ordre d’introduction n’a pas d’importance. • Séquence suggérée : céréales, légumes, fruits, viande. • Il est préférable d’offrir les aliments solides avant le lait pour encourager le bébé. Vers 6 mois, il faut limiter la quantité de lait offerte au bébé à 600 ml. • On introduit : o jus de fruits à 6 mois, 120 ml/jour. o Jaune d’œuf à 7-8 mois (si allergie familiale → 18 mois) o Blanc d’œuf à 12 mois (si allergie familiale → 18 mois) • Ne pas ajouter du sucre ou du sel pendant la première année de vie. • Texture : o 6 mois : purées o 8 mois : purées plus épaisses o 8-12 mois : aliments hachés finement, râpés, puis en petites lanières et en petits morceaux tendres. • Vers l’âge de 1 an, le bébé devrait manger la même variété alimentaire que sa famille. • Si ATCD d’allergies familiales, pas d’arachides, miel, noix ou fruits de mer avant 3 ans • Fruits des champs pas avant 1 an, si oui il faut que ça soit cuit pour dénaturer les protéines) • Pas de miel avant 1 an : risque de botulisme • Donner des suppléments de fer au bébé allaité de 6 mois qui ne prend pas beaucoup d’aliments solides. L’introduction précoce des aliments solides a été associée à un risque accru de problèmes atopiques. |
|
|
|
Is scoliosis screening recommended?
|
The canadian task forces on Preventive Health Care do NOT recommend routine screening using the forward bend test.
Cohort studies indicate that the forward bend test has poor sensitivity for identifying pathological curves. |
|
|
|
How much weight should a newborn gain per day?
|
20-30 g/day
"1 oz. per day except on sunday" |
|
|
|
Formula to estimate weight of a child > 1 year
|
Age x 2 + 8
|
|
|
|
Head circumference mnemonic
|
Remember 3, 9 and multiples of 5
Newborn 35 cm 3 months 40 cm 9 months 45 cm 3 years 50 cm 9 years 55 cm |
|
|
|
Normal evolution of newborn weight
|
Weight loss up to 10% in first 7 days
Neonate should regain weight by 10 days 2 x birth weight by 4-5 MO 3 x birth weight by 1 year 4 x birth weight by 2 years |
|
|
|
Normal evolution of newborn Length/Height
|
Growth :
- 25 cm in 1st year - 12 cm in 2nd year - 8 cm in 3rd year - 4/7 cm/year until puberty (1/2 adult height at 2 years) Measure supine length until 2 years of age, then measure standing height. |
|
|
|
Development of dentition
|
1) primary dentition (20 teeth)
- first tooth at 5-9 months (lower incisor), then 1 per month until 20 teeth. - 6-8 central teeth by 1 year 2) secondary dentition (32 teeth) - first adult tooth is 1st molar at 6 years, then lower incisors - 2nd molars at 12 years, 3rd molars at 18 years |
|
|
|
Failure to thrive patterns
|
Decreased Wt, Normal Ht, Normal HC
- Insufficient intake - Decreased intake - Hypermetabolic state - Increased losses Decreased Wt, Decreased Ht, Normal HC - Structural dystrophies - Endocrine disorder - Constitutional growth delay - Short familial stature Decrease Wt, Decreased Ht, Decreased HC - Intra-uterine insult - Genetic abnormality |
|
|
|
Definition of failure to thrive
|
Child 2 years or younger with one of these criteria :
1) Weight < 3rd percentile 2) Weight falls across two major percentile curves 3) Weight < 80% of expected weight for height and age (ideal weight) |
|
|
|
Energy requirements
|
0-10 Kg : 100 cal/kg/day
0-20 Kg : 1000 cal + 50 cal/kg/day for each Kg > 10 >20 Kg : 1500 cal + 20 cal/kg/day for each Kg > 20 |
|
|
|
History elements to retrieve for failure to thrive
|
Duration of problem and growth history
Detailed dietary and feeding history, appetite, behaviour before and after feeds, bowel habits. Pregnancy, birth and postpartum history Developmental and medical history (including medications) Social and family history (parental height, weight, growth pattern, diseases) Assess 4 areas of functioning : child's temperament, child-parent interaction, feeding behaviour and parental psychosocial stressors. |
|
|
|
Physical exam for failure to thrive
|
Height, Weight, Head Circumference, Arm span, upper to lower segment ratio
Assessment of nutritional status, dysmorphism, Tanner stages, evidence of chronic disease Observation of a feeding session and parent-child interaction Signs of abuse or neglect Observe adipose tissue, muscles and presence of edema. |
|
|
|
Investigations for failure to thrive
|
CBC, Blood smear, ESR
Electrolytes, Urea, Urinalysis T4, TSH Bone age X-Ray (Left wrist) Karyotype in all short girls and boys where appropriate Other tests according to interview and physical exam : renal or liver function, venous blood gases, ferritin, immunoglobulins, sweat chloride, fecal fat. |
|
|
|
Causes of failure to thrive
|
Organic causes
- Inability to feed * insufficient breast milk production * poor retention (GERD, vomiting) * CNS, neuromuscular, mechanical problems with swallowing, sucking * anorexia (associated with chronic disease) - Inadequate absorption * malabsorption : celiac disease, cystic fibrosis, pancreatic insufficiency * loss from the GI tract : chronic diarrhea, vomiting - Inadequate utilization of nutrients * renal losses : tubular disorders * inborn errors of metabolism * endocrine : type I diabetes, diabetes insipidus, hypopituitarism, congenital hypothyroidism - Increased energy requirements * pulmonary disease : CF * cardiac disease * endocrine : hyperthyroidism, DI, hypopituitarism * malignancies * chronic infections * inflammatory : SLE Non-Organic causes - Malnutrition - Inadequate nutrition - Poor feeding technique - Errors in making formula |
|
|
|
Clinical signs of failure to thrive
|
SMALL KID
Subcutaneous fat loss Muscle atrophy Alopecia Lethargy Lagging behind normal Kwashiorkor Infection (recurrent) Dermatitis |
|
|
|
Signs of non organic failure to thrive
|
Children are picky eaters with poor emotional support at home or poor temperament
May have delayed psychomotor, language and personal/social development, Emotional deprivation, poor parent-child interaction, dysfunctional home Child abuse or neglect Parental psychosocial stress, personal history of suffering abuse or neglect |
|
|
|
Diagnosis of obesity
|
Weight > 95th percentile
BMI > 30 Weight > 20% greater than ideal weight |
|
|
|
Risk factors for obesity
|
Prevalence of obesity in Canada has tripled.
It is caused by a chronically positive energy balance. Risk factors : - if 1 parent is obese : 40% chance of obese child - if 2 parents are obese : 80% chance of obese child |
|
|
|
Secondary causes of obesity
|
Organic causes of obesity are rare (< 5%)
Endocrine - Hypothyroidism - Cushing syndrome Genetic - Prader Willi - Carpenter - Turner |
|
|
|
Complications of obesity
|
Association with : HTN, DLPD, slipped capital femoral epiphysis, Type 2 diabetes, asthma, obstructive sleep apnea.
Boys : gynecomastia Girls : polycystic ovarian syndrome, early menarche, irregular menses Psychosocial : teasing, decreased self esteem, unhealthy coping mechanisms, depression ___________________________ N.B. Childhood obesity is an unreliable predictor of adult obesity. - unless > 180% of ideal weight - however, 70% of obese adolescents become obese adults |
|
|
|
Management of obesity
|
Encouragement and reassurance; engagement of entire family
Diet : qualitative changes; do not encourage weight loss but allow for linear growth to catch up with weight; special diets used by adults are not encouraged. Evidence against very low calorie diets for pre adolescents Behaviour modification : increase activity, change eating habits / meal patterns Surgery and pharmacotherapy are not used in children |
|
|
|
Result of the geographic and demographic variation in the prevalence of overweight canadian children.
|
The prevalence of childhood obesity is increasing in all areas of Canada, although more so in Atlantic Canada.
|
|
|
|
Diagnosis of colic
|
Rule of 3's :
Unexplained paroxysms of irritability and crying for : > 3hours > 3 days/week > 3 weeks in an otherwise healthy, well-fed baby. They usually occur between 10 days to 3 months. peak : 6-8 weeks. Child cries, pulls up legs and passes gas soon after feeding. |
|
|
|
Etiology of colics
|
Generally regarded as a lag in the development of normal peristaltic movement in GI tract, other theories suggest a lack of self soothing mechanisms.
Other reasons why babies cry : wet, hunger or gas pains, too hot or cold, overstimulated, need to suck or be held. |
|
|
|
Management of colic
|
Parental relief, rest and reassurance
Hold baby, soother, car ride, music, vacuum, check diaper Medications (Ovol drops, gripe water) of no proven benefit if breast feeding : elimination of cow's milk protein from mother's diet (effective in very small percentage of cases) Try casein hydrosylates formula (Nutramigen) |
|
|
|
What is the leading cause of death in children > 1 year of age
|
Injuries
- motor vehicle crashes - burns - drowning - falls - choking - infanticide |
|
|
|
Milk Caries cause and prevention
|
Decay of superior front teeth and back molars in first 4 years of life
Often occur in children put to bed with a bottle of milk or juice Can also be causes by breast feeding (especially prolonged night feeds) Prevention : - no bottle at bedtime (unless plain water) - use water as thirst quenchers during the day, do not sweeten pacifier - can clean teeth with soft damp cloth or toothbrush and water - avoid fluoridated toothpaste until able to spit (> 3 years) because of fluorosis risk - Canadian Dental Association recommends assessment by dentist 6 months after eruption of first tooth, or by 1 year of age. |
|
|
|
Definition of sudden infant death syndrome
|
Sudden and unexpected death of an infant < 12 months of age in which the cause of death cannot be found by history, examination or a thorough postmortem and death scene investigation.
|
|
|
|
Risk Factors for sudden infant death syndrome
|
Risk Factors :
- prematurity - smoking in household - minorities - socially disadvantaged More common in children placed in prone position Increase in deaths during peak RSV season Most deaths occur between midnight and 8 am. In full term infants, peak incidence is 2-4 months, 95% of cases occur by 6 months |
|
|
|
Prevention of sudden infant death syndrome
|
Place infant on back, not in prone position when sleeping.
Allow supervised play time daily in prone position Alarms / other monitors not recommended - increase anxiety and do not prevent life threatening events Avoid overheating and overdressing Appropriate infant bedding No smoking Pacifiers appear to have a protective effect; do not reinsert if falls out |
|
|
|
Age for toilet training and signs of toilet readiness
|
25% by 2 years old, 98% by 3 years old have daytime bladder control.
Signs of toilet readiness : - ambulating independently - stable on potty - desire to be independent or to please caregivers - sufficient expressive and receptive language skills - can stay dry for several hours |
|
|
|
Definition of enuresis
|
Involuntary urinary incontinence by day and/or night (typically 5-6 yrs old)
Wetting at least twice a week for at least 3 consecutive months or causes significant distress to the child. Treatment should not be considered until 6 years of age. high rate of spontaneous cure. |
|
|
|
Types of enuresis
|
Primary nocturnal enuresis = never had continence > 6 months
Secondary enuresis = already had continence for > 6 months Diurnal euresis |
|
|
|
Primary nocturnal enuresis
|
Wet only at night during sleep, can be normal up to age 6.
Prevalence : 10% of 6 year olds, 3% of 12 year olds, 1% of 18 year olds Primary enuresis if no episode of continence for > 6 months. Developmental disorder or maturational lag in bladder control while asleep. More common in boys, family history common |
|
|
|
Treatment of primary nocturnal enuresis
|
1- Time and reassurance (20% resolve spontaneously each year)
2- Behaviour modification : limiting nighttime fluids, voiding prior to sleep, engaging child using rewards, bladder retention exercises, scheduled toileting. 3- Conditioning : "wet" alarm wakes child upon voiding (70% success rate) 4- Medications (2nd line therapy) 4.1 DDAVP by nasal spray or oral tablets (high relapse rate) 4.2 Oxybutynin (Ditropan) - anticholinergic 4.3 Imipramine (Tofranil) - tricyclique |
|
|
|
Secondary enuresis
|
Develops after child has sustained period of bladder control (6 months or more)
|
|
|
|
Causes of primary diurnal enuresis
|
Neurogenic bladder (cerebral palsy, spina bifida)
Congenital anomalies (ectopic ureter, posterior uretral valve, cloacal anomalies, exstrophiesépispadias) Diabetes Diabetes insipidus |
|
|
|
Causes of secondary diurnal enuresis
|
Bladder instability
Procrastination maneuvers Micturition retention Urinary infection Constipation Coalescence of labia minora Vaginal reflux Bladder-sphincterian asynergy Laugh or stress incontinence Treatment depends on cause |
|
|
|
Important investigations for enuresis.
|
Primary nocturnal enuresis
- urinalysis, urine culture, urine sediment Diurnal enuresis - Kidneys ultrasound, if not normal, proceed to mictionnal cystography - If history is suggestive of neurologic damage, MRI is indicated |
|
|
|
Definition of encopresis
|
Fecal incontinence in a child > 4 years old at least once per month for 3 months.
Fecal encopresis can be with or without fecal retention |
|
|
|
Causes of encopresis
|
Usually associated with chronic constipation.
Must exclude medical causes : Hirschprung disease, hypothyroidism, hypercalcemia, spinal cord lesions, anorectal malformations. |
|
|
|
Causes of retentive encopresis
|
Anal fissure
Disturbed parent-child relationship, coercive toilet training, social stressors (child tries to retain his stool as much as possible to please, but it comes to a point where he gets encopresis) |
|
|
|
Signs of retentive encopresis
|
Child tries to hold stool in order to achieve continence. He develops constipation leading to fecal impaction. Maneuveres to withhold going to bathroom. Distresses by symptoms --> soiling of clothes.
Physical exam : DRE = large fecal mass in rectal vault |
|
|
|
Treatment of retentive encopresis
|
1- Complete clean out of bowel
- enemas and suppositories 2- Maintenance of regular bowel movements - stool softeners (colace, lactulose ...) - diet modification - toilet schedule and positive reinforcement 3- Assessment and guidance regarding psychosocial stressors 4- Behavioural modification |
|
|
|
Complications of retentive encopresis
|
Toxic megacolon
Bowel perforation |
|
|
|
What is a breath holding spell?
|
Child is provoked (usually by anger, injury or fear), starts to cry and then becomes silent. Spell resolves spontaneously or the child may lose consciousness; rarely progresses to seizures.
2 Types : - cyanotic (more common), usually associated with anger or frustration - pallid, usually associated with pain or surprise Treatment : - behavioural : help child control response to frustration and avoid drawing attention to spell. |
|
|
|
Characteristics of innocent heart murmur
|
Ejectional systolic murmur
< 3/6 Physiologic splitting of S2 No symptoms No added sounds Murmur varies with change of position No palpable thrill no structural or ECG changes |
|
|
|
Innocent heat murmurs
|
1) Peripheral pulmonic stenosis (neonates, usually disappears by 3-6 MO) - radiates to back and axilla
2) Still's murmur (3-6 years) - vibratory, lower left sternal border or apex 3) Venous hum (3-6 years) - infraclavicular hum, continuous, R>L 4) Pulmonary ejection (8-14 years) - Soft, blowing, upper left sternal border 5) Supraclavicular arterial bruit (any age) - Low intensity above clavicles |
|
|
|
Prenatal circulation (shunts and circulation)
|
Shunts
- Oxygenated blood 1) Ductus venosus : connecting between umbilical vein and IVC 2) Foramen Ovale : connecting right atria with left atria - Deoxygenated blood 1) Ductus arteriosus : connecting pulmonary artery with aorta ___________________________________________________________________ Circulation - Placenta (O2) > umbilical vein > ductus venosus > IVC > RA > Foramen ovale > LA > LV > aorta > brain/myocardium/upper extremities - Deoxygenated blood returns via SVC to RA > 1/3 of blood entering RA flows to RV > pulmonary arteries > ductus arteriosus > aorta > systemic circulation > placenta for reoxygenation |
|
|
|
What happens to the prenatal circulation at birth?
|
At birth :
1) With first breath, lungs open up and pulmonary resistance decreases allowing pulmonic blood flow. 2) With separation of placenta, systemic circulation becomes a high resistance system. 3) With closure of the fetal shunts and changes in pulmonic / systemic resistance, infant circulation assumes normal adult flow 4) Increasing pulmonic flow increases left atrial pressures leading to foramen ovale closure 5) Increased oxygen concentration in blood after first breath leads to decreased prostglandins leading to closure of the ductus arteriosus. 6) As the umbilical cord is clamped, the umbilical vein closes, systemic vascular resistance increases and the ductus venosus closes. In Resume : - closure of foramen ovale - closure of ductus arteriosus - closure of ductus venosus - increase in systemic resistance |
|
|
|
Risk factors for congenital heart disease
|
Prematurity
Teratogenicity : - Maternal pregnancy infections : rubeola (VSD, PDA, pulmonary artery stenosis) + TORCH - Alcohol during pregnancy (VSD, ASD) - Medications during pregnancy : isotrétinoine (TOF), lithium (Ebstein), Warfarin, thalidomide, Valproate, Phenytoin - Auto-immune maternal disease : LSE, RA - Maternal diabetes (2% to 3%) - Maternal phenycetonuria Genetic syndromes: - Down syndrome (atrioventricular septal defect) - Turner syndrome ( usually coarctation of the aorta) - Kartagener's - Marfan - Osteogenesis imperfecta - CHARGE association - DiGeorges - VATER association |
|
|
|
If 1 infant has Cardiac malformation, what is the risk for his borthers/sisters?
|
2% to 6%
|
|
|
|
CHARGE syndrome
|
The following are the signs that were originally identified in children with this syndrome, but these features alone are no longer used in official diagnosis.
C - Coloboma of the eye, central nervous system anomalies H - Heart defects A - Atresia of the choanae R - Retardation of growth and/or development G - Genital and/or urinary defects (Hypogonadism) E - Ear anomalies and/or deafness The most common genital condition associated with CHARGE syndrome is undescended testicles, or cryptorchidism. Another commonly associated genital condition is hypospadias. |
|
|
|
Possible manifestations of cardiac malformations
|
Infant :
- heart murmur - respiratory distress - cyanosis - FTT - CHF - frequent respiratory infections - feeding problems Older child : - exertion dyspnea - cyanosis - squatting - clubbing - syncope - FTT |
|
|
|
Investigations for cardiac malformations
|
ECG
CXR Heart Ultrasound |
|
|
|
Characteristic X-Ray findings in congenital heart disease :
- Tetralogy of Fallot (TOF) - Transposition of great arteries - Total anomalous pulmonary venous return |
- Tetralogy of Fallot (TOF) : Boot shaped heart
- Transposition of great arteries : Egg shaped heart - Total anomalous pulmonary venous return : Snowman heart |
|
|
|
Congenital Cardiac Malformations
|
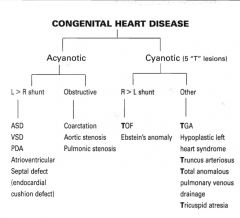
|
|
|
|
Common congenital heart disease pictures.
- TOF - TOGA - Coarctation of the aorta - Patent ductus arteriosus |
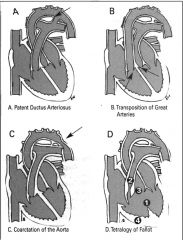
|
|
|
|
Minimal required concentration of deoxygenated hemoglobin to see cyanosis.
|
Hb > 3 g/dL
|
|
|
|
Acyanotic congenital heart diseases
|
L > R shunt
- ASD - VSD - Patent ductus arteriosus - AVSD Obstructive - Coarctation - Aortic stenosis - Pulmonic stenosis |
|
|
|
Cyanotic congenital heart diseases
|
R > L shunt
- Tetralogy of Fallot - Ebstein's anomaly Other - Transposition of great arteries - Hypoplastic left heart syndrome - Truncus arteriosus - Total anomalous pulmonary venous drainage - Tricuspid atresia |
|
|
|
5 T's of congenital heart disease
|
Tetralogy of Fallot
Transposition of great arteries Tricuspid atresia Total anomalous venous drainage Truncus arteriosus |
|
|
|
Complications of L > R shunts
|
Pulmonary vascular disease
Right ventricular hypertension and hypertrophy R > L shunts (pulmonary pressure too great, so blood preferably passes through the shunt) |
|
|
|
Types of atrial septal defect
|
Ostium primum (common in Down syndrome)
Ostium secundum (most common type 50-70%) Sinus venosus (defect located at entry of superior vena cava into right atrium) |
|
|
|
Risks of ASD
|
Natural history : 80%-100% spontaneous closure rate if diameter < 8 mm
If ASD remains patent, there is an increased risk of CHF and pulmonary hypertension during the adult life. |
|
|
|
Treatment of ASD
|
elective surgery or catheter closure between 2-5 years of age
|
|
|
|
Presentation of ASD
|
10% of congenital heart malformations
Usually asymptomatic. Can present with Heart failure or FTT or exertion dyspnea. P/E : Doubled S2 + systolic ejectionnal murmur |
|
|
|
Investigations of ASD
|
ECG : RBBB, Right axis deviation, mild RVH
CXR : increased pulmonary vasculature + cardiomegaly |
|
|
|
Atrioventricular canal picture
|
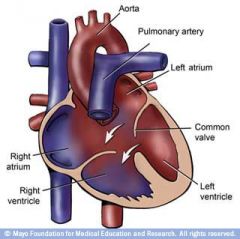
|
|
|
|
Presentation of ventricular septal defect
|
30% of congenital heart malformations
Small VSD : - No symptoms usually, resolves spontaneously, systolic murmur, ECG + CXR normal. Moderate to large VSD : - Natural history : pulmonary hypertension, chronic heart failure by 2 months of age + FTT. - Systolic murmur, if very large no murmur |
|
|
|
Types of VSD
|
Muscular
Membranous Sub arterial Atrioventricular canal |
|
|
|
Investigations of VSD
|
CXR : increased vasculature + cardiomegaly
ECG : LVH, LAH, RVH Echo : Identifies the defect and its location |
|
|
|
Treatment of varicella
|
- Supportive (hydration, acetaminophen, antipruritics, AVOID salicylates)
- Proper hygiene, discourage scratching - Acyclovir for severe disease, immunocompromised patients, neonates - Avoid contact with others until lesions are dry and crusted and no new ones are appearing |
|
|
|
Management of a neonate born to mothers who develop varicella from 5 days before to 2 days after delivery
|
Must administer VZV immunoglobulins and follow for signs of infection/sepsis, consider staring acyclovir
|
|
|
|
Maternal infection with Varicella complications
|
Maternal infection in first or early second trimester (<2%) can cause congenital varicella syndrome (low birth, CNS abnormalities, digit/limb abnormalities, cutaneous scarring, eye defects).
Maternal infection 5 days before to 2 days after delivery can lead to severe varicella of neonate |
|
|
|
Roseola (roséole)
|
HHV-6
Incubation : 5-15 days; infectivity and spread : unknown Typically affects children < 3 years Clinical presentation - High fever (>39.5) lasting 3-5 days, cough, respiratory symptoms, nasal congestion - Pharynx, tonsils and tympanic membranes are erythematous - Cervical, posterior cervical lymphadenopathy, bulging anterior fontanelle (if CNS involvement) - Fever ceases before rash appears + Pink non pruritic macules and maculopapules + Macules coalesce and disappear in 1-2 days Treatment - Supportive Complications - Febrile seizures - Encephalitis |
|
|
|
Measles (rougeole)
|
Morbillivirus
Incubation : 10-14 days; infectivity : 4 days pre-rash, spread by airborne route Clinical presentation : - prodrome "3 C's" : cough, coryza, cojunctivitis, fever, eyelid edema - koplik spots (1-2 days before and after rash) : small white papules on red base on buccal mucosa - maculopapular rash spreads over face and hariline spreading in a descending fashion over the body over 3 days. Diagnosis : - Clinical examination and positive serology for measles IgM Treatment : - supportive and symptomatic - prophylactic immunoglobulin to prevent disease if administered within 6 days of exposure - vitamin A supplementation in selected children |
|
|
|
Koplik spots
|
Seen in measles (rubeolla)
koplik spots (1-2 days before and after rash) : small white papules on red base on buccal mucosa |
|
|
|
Complications of measles
|
Secondary bacterial infection (laryngotracheobronchitis, otitis media, sinusitis), bronchopneumonia, croup.
Encephalitis Subacute sclerosing panencephalitis : slow measles virus infection of brain manifesting years later, characterized by progressive cerebral deterioration with myoclonic jerks, fatal within 6-12 months. |
|
|
|
Mumps (Oreillons)
|
Paramyxovirus
Incubation : 12-25 days; infectivity : 7 days pre-parotitis to 7 days post-parotitis, spread by droplets Clinical presentation - Fever, headache, parotitis (bilateral, pushes earlobes up and out), myalgia, malaise - 30%-40% of cases are subclinical with minimal symptoms Treatment - Supportive Diagnosis - Urine or saliva for viral serology Complications - meningoencephalomyelitis : over 10% of patients with parotitis - orchitis, epididymitis, infertility - pancreatitis - other : ocular complications, thyroiditis, hearing impairment, myocarditis, arthritis, thrombocytopenia, cerebellar ataxia, glomerulonephritis |
|
|
|
Congenital rubella syndrome
|
- Mother infected in first 4 months of pregnancy (highest risk)
- Infection in utero, failure of rubella vaccine < 5% Presentation - Cataracts/congenital galucoma, congenital heart disease, hearing impairment, purpura, hepatosplenomegaly, jaundice, microcephaly, developmental delay, radiolucent bone diseae Prevention - Routine childhood immunization - Assure immunity of women of childbearing age with vaccination |
|
|
|
Reye syndrome
|
Acute hepatic encephalopathy and noninflammatory fatty infiltration of liver and kidney
Associated with aspirin ingestion by children with varicella or influenza infection Diagnosis by liver biopsy 40% mortality rate Clinical presentation : - vomiting - hyperventilation, tachycardia, decerebrate posturing - respiratory failure - agitated delirium, coma, death Treatment : - should be tailored on severity of presentation - IV glucose (to counteract effects of glycogen depletion) - Fluid restriction, mannitol (if cerebral edema) - Prevention : avoid aspirin with viral illness |
|
|
|
Erythema infectiosum
|
Fifth disease or Parvovirus B19
Incubation : 4-14 days; infectivity : prior to onset of rash Clinical presentation : - initial 7-10 days : flu-like illness with fever - day 10-17 : rash appears + raised maculopapular lesions on cheeks (slapped cheek appearance), forehead chin, circumoral sparing + warm, nontender, may be pruritic, may also appear on extensor surfaces, trunk neck - days to weeks : rash fades, may reappear with local irritation (heat, sunlight) Treatment - supportive - blood transfusions for some with aplastic crisis Complications - arthritis, vasculitis - infection during pregnancy may lead to fetal hydrops, fetal loss - aplastic crisis : reticulocytopenia occurs for 1 week during illness, unnoticed in normal individuals, but severe anemia in patients with chronic hemolytic anemia |
|
|
|
Definition of urinary tract infection in children
|
Urine specimen with >100 000 colonies/ml of a single organism
|
|
|
|
Common organisms in UTI
|
Klebsiella
E. Coli Enterococcus Proteus S. Saprophyticus Pseudomonas |
|
|
|
Presentation and diagnosis of UTI in children
|
Cystitis : dysuria, frequency, suprapubic pain, incontinence, malodorous urine
Pyelonephritis : abdominal or flank pain, fever, malaise, No/Vo |
|
|
|
Screening and diagnosis of UTIs in children
|
Screening
- reactive dipstick - urinary microscopy Diagnosis method - Supra-pubic aspiration (Sp 99%) - Transurethral catheter (Sp 85%) Diagnosis - Newborn : positive urinary microscopy with one positive culture (supra-pubic aspiration) - Child : positive urinary sediment with 2 positive cultures (sample) or 1 positive sample (supra-pubic aspiration) - Adolescent : positive urinary sediment with 2 positive cultures with the same bacteria If systemically ill : CBC, electrolytes, Cr, BUN, blood cultures |
|
|
|
Radiologic evaluation for UTIs
|
U/S to assess for renal growth, hydronephrosis, or structural anomalies and voiding cystorurethrography to assess for VUR for all children < 2 years presenting with febrile UTI
Renal scintigraphy if urethrogram abnormal or history of pyelonephritis to assess for renal scarring Mag3-Furosemide scintigraphy to distinguish between obstructive and non obstructive hydronephrosis |
|
|
|
Treatment of UTIs in children
|
Encourage fluid intake
Child < 3 months - IV Ampicillin + Gentamycin x 10-14 days, switch to PO when fever goes down - Gentamycin dosage - 2nd line : Cefotaxim (no enteroc. protection) Child 3 months - 6 years - Fever + dehydration + toxic + IV ampicillin + gentamycin, switch to PO when fever goes down x 10-14 days + Appropriate ATB after culture results + Gentamycin dosage - Fever only + IV ampicillin + gentamycin, switch to PO when fever goes down x 10-14 days + Appropriate ATB after culture results + Gentamycin dosage - No symptoms + Augmentin PO x 10 days + 2nd line Cefixime Child > 6 years - Pyelonephritis + if really ill : same as 3mo-6yrs + 6-14 years : augmentin or cefixime PO + 15-18 years : Cipro or bactrim - Cystitis + Cephalexine x 3-4 days (10 days if GU anomalies) + 2nd line Bactrim |
|
|
|
Complications of UTIs
|
Bacteremia
Abscess Local nephritis Renal scarring Hypertension CRF |
|
|
|
When is circumcision indicated?
|
Circumcision is indicated when the newborn has recurrent urinary infections before 6 months of age.
|
|
|
|
Is prophylaxis recommended after first UTI in children?
|
No
Bactrim is recommended for all children with reflux, awaiting investigations and/or > 3 UTIs/year, Trimethoprim alone if < 2 Mo |
|
|
|
Management of urinary voiding dysfunction
|
Intensive oral hydration
Miction every 2-3 hours Miction in 2 times Have a miction calendar At least 1 bowel movement per day |
|
|
|
Causes of hematuria in children
|
|
|
|
|
Evaluation of persistent proteinuria in children
|
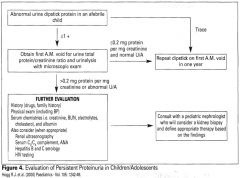
|
|
|
|
Causes of coloured urine with negative dipstick
|
URINE BLEAD
Urates Rifampin Ibuprofen Nitrofurantoin Exogenous (food colouring) Beets Lead |
|
|
|
False positive dipstick (positive dipstick, but no RBCs)
|
Myoglobinuria dur to rhabdomyolysis
Hemoglobinuria due to intravascular hemolysis or coagulation |
|
|
|
Alport syndrome
|
Alport syndrome is a genetic disorder characterized by glomerulonephritis, endstage kidney disease, and hearing loss.
|
|
|
|
Causes of persistent proteinuria in children
|
- Orthostatic
- Glomerular - Tubulointerstitial (Fanconi, ATN) - Structural abnormalities of urinary tract |
|
|
|
Causes of acute glomerulonephritis in children
|
|
|
|
|
Causes of acute glomerulonephritis in children
|
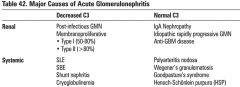
|
|
|
|
Causes of HTN in children (by age)
|

|
|
|
|
Normal values of HTN in children (by age)
|

|
|
|
|
Investigations for hypertension in children
|

|
|
|
|
Management of HTN in children
|

|
|
|
|
Hemolytic uremic syndrome
- Epidemiology - Pathophysiology - Clinical presentation - Investigations - Treatment - Prognosis |
Epidemiology
- most common cause of ARF in children - most common cause from 6 months to 4 years of age Pathophysiology - E. Coli O157:H7 verotoxin or shiga toxin + toxin binds, invades and destroys colonic epithelial cells, causing bloody diarrhea + toxin enters the systemic circulation, attaches and injures endothelial cells causing a release of endothelial products + formation of platelet / fibrin thrombi in multiple organ systems (renal, pancreas, brain) resulting in thrombocytopenia + RBCs are forced through occluded vessels resulting in fragmented RBC (schistocytes) and removed by reticuloendothelial system (hemolytic anemia) + other, rare forms of HUS in childhood are due to bacteria (S. Pneumoniae), viruses, familial inheritance, or drugs. Clinical presentation - triad : ARF, thrombocytopenia, microangiopathic hemolytic anemia - initial presentation of abdominal pain and diarrhea, followed by bloody diarrhea - within 5-7 days the patient begins to show signs of anemia, thrombocytopenia and renal failure + Hx : weakness, lethargy, oliguria + Physical exam : pallor, jaundice, edema, petechiae, hypertension Investigations - CBC, platelets, blood smear, Urinalysis, BUN, creatinine, stool culture and shigella toxin Treatment - Supportive : nutritional + rehydration - Monitor electrolytes, dialysis if electrolyte abnormality cannot be corrected - PRBC for symptomatic anemia - Steroids not helpful; antibiotics not indicated Prognosis : 5-10% mortality, 10-30% kidney damage |
|
|
|
HUS triad
|
ARF
Microangiopathic hemolytic anemia Thrombocytopenia |
|
|
|
Nephritic syndrome characteristics
|
Hematuria
Hypertension Proteinuria (<50mg/kg/d) Azotemia RBC casts Oliguria |
|
|
|
Post-streptococcal GN
- Risk factors - Pathophysiology - Diagnosis - Prognosis - Management |
Risk factors
- most common in children, aged 4 to 8 years old M>F - occurs 1-3 weeks following group A beta hemolytic streptococcal infection of skin or throat Pathophysiology - antigen-antibody mediated complement activation - diffuse proliferative glomerulonephritis Diagnosis - elevated serum antibody titres against strep antigens (ASOT) Prognosis - 95% of children recover completely within 1-2 weeks - 5-10% have persistent hematuria Management - symptomatic treatment : fluid restriction, anithypertensives, diuretics - in severe cases : hemodialysis or peritoneal dialysis - eradication of infection (Penicillin or Erythromycin) |
|
|
|
Etiology of nephrotic syndrome
|
Primary nephrotic syndrome
- Minimal change disease - Membranous glomerulonephritis - Focal segmental glomerular sclerosis - Membranoproliferative glomerulonephritis Secondary nephrotic syndrome - vasculitis - infections (HBV, HCV, Syphilis, HIV) - medications (Captopril, Penicillamine, NSAIDs, anticonvulsants) - malignancy - hereditary (sickle cell disease, Alport syndrome) - metabolic, inflammatory (Lupus, rheumatoid arthritis) |
|
|
|
Complications of nephrotic syndrome
|
- Risk of infections
- Hypercoagulability due to decreased intravascular volume and antithrombin III depletion (PE, renal vein thrombosis) - Side effects of drugs (diuretics, steroids, immunosuppressants) - Hypotension, shock, renal failure |
|
|
|
Management of nephrotic syndrome
|
- Salt and water restriction, diuretic may be required
- Optimal nutrition, including high quality protein - Daily weights to assess therapeutic progress - Pneumococcal vaccine after remission - Initial treatment of MCD + Oral prednisone 60 mg/m2/day in divided doses for up to 12 weeks + a negative tuberculin skin test should be performer before starting steroid medications + a measurable decrease in protein excretion may take at least 7 to 10 days following initiation of treatment and proteinuria clears by third week of oral prednisone + up to 2/3 of patients experience relapses - If unresponsive to steroids, frequent relapses, or steroid resistant + consider renal biopsy or treat with cytotoxic agent (cyclophosphamide) .... |
|
|
|
Paediatric blood pressure calculation
|
sBP = age x 2 + 90
dBP = 2/3 sBP |
|
|
|
Malignant conditions associated with retinoblastoma?
|
Osteosarcoma
Less frequently : - glioblastoma - malignant melanoma - squamous cell carcinoma |
|
|
|
Gastrografin enema
|
A gastrografin enema has a dual diagnosis/therapeutic role in paediatric surgery for meconium ileus
|
|
|
|
Which vessels are damaged in shaken baby syndrome?
|
Bridging veins of the skull.
|
|
|
|
Marfan syndrome
|
Connective tissue disease with autosomal dominant transmission
Cardinal features of the disease include : - Tall stature - Ectopia lentis - Mitral valve prolapse - Aortic root dilatation - Aortic dissection Early intervention with with beta adrenergic blocking agents and careful echocardiography screening may help slow aortic dilatation and allow aortic root replacement therapy before dissection occurs |
|
|
|
Dacryostenosis
|
Congenital nasolacrimal duct obstruction
It occurs in at least 6% of all newborns. It is characterized by unilateral tearing and yellow crusting of the eye each morning, sometimes so thick, that the infant cannot open the eyelids. Parents are advised to massage the inner canthus of the eye twice daily to encourage resolution of the obstruction. Resolution occurs by 1 year of age in 90% of infants. |
|
|
|
Which tumour is associated with aniridia?
|
Wilms tumour (renal)
|
|
|
|
Neuroblastoma
|
Neruoblastoma arises from neural crest tissu and occurs in infancy. It is the most common solid tumour in children other than brain tumours. 85% of neuroblastoma cases are diagnosed in children < 2 years. The clinical presentation varies with the tumour's location.
Common sign and symptoms include : - asymptomatic abdominal mass - Horner syndrome - Persistent cough - Superior vena cava syndrome - Bone pain - Cord compression - Subcutaneous nodules ("blueberry muffin" lesions) - Hypertension - Opsoclonus ("dancing eyes") - Myoclonus ("dancing feet") |
|
|
|
Williams syndrome
|
Williams syndrome is a deletion of the 7q chromosome, causing short stature, hypercalcemia or hypercalciuria in infancy; developmental delay, dysmorphic features, overly friendly personality and supravalvular aortic stenosis.
|
|
|
|
Hartnup disease
|
Autosomal recessive condition that produces a neutral aminoaciduria with increased renal clearance of amino acids.
This is accompanied by malabsorption of other amino acids, notably tryptophan, but also phenylalanine and methionine. The resulting tryptophan deficiency produces pellagra-like symptoms with photosensitive skin lesions, ataxia and neuropsychiatric disturbances. |
|
|
|
Blood smear findings of abetalipoprteinemia
|
Acanthocytes
|
|
|
|
Most common tumours in children
|
Medulloblastoma
Ependymoma Pilocytic astrocytoma |
|
|
|
Coxsackie infection in children
|
Herpangina : dysphagia + vesicles on the anterior tonsila and palate.
Hand-foot-and-mouth disease is also caused by coxsackie virus and presents with small vesicles in the mouth, but there are also vesicles on the hands and feet. Commonly, a maculopapular rash is present. |
|
|
|
Talipes equinovarus
|
Most common form of clubfoot abnormality
Equinus : Permanent extension of the foot so that only the ball rests on the ground. Varus : permanent inversion of the foot, so that only the outer side of the sole touches the ground. |
|
|
|
Talipes calcaneovalgus
|
Flat and dorsiflexed foot.
|
|
|
|
Bleeding from the ear after head injury is pathognomonic of???
|
Temporal bone fracture
|
|
|
|
Acetaminophen
|
10-15 mg/kg/dose PO/PR q4-6h prn
|
|
|
|
Ibuprofen
|
5-10 mg/kg/dose PO Q6-8H
|
|
|
|
Dexamethasone
|
0.6mg/kg IV x 1 or 1mg/kg PO x1 (croup)
0.3 mg/kg/day PO (asthma) |
|
|
|
Fluticasone (Flovent)
|
Moderate dose : 25-500 ug/day divided bid
High dose : >500 ug/day divided bid |
|
|
|
Iron
|
6 mg/kg/day elemental iron bid-tid
|
|
|
|
Ventolin
|
001-0.03 ml/kg/dose in 3 ml normal saline via nebulizer q 1/2-4 H prn
|
|
|
|
Differential diagnosis of stridor
|
Tracheitis
Laryngotracheobronchiolitis Croup Foreign body aspiration Sub-glottic stenosis (congenital or iatrogenic) Laryngomalacia - Tracheomlacia (collapse of epiglottis cartilage on inspiration) |
|
|
|
Dose of racemic epinephrine for croup
|
Nebulized
1-3 doses Q1-2H |
|
|
|
Differential diagnosis of wheezing
|
Common
- Asthma - Bronchiolitis - Recurrent aspiration - Pneumonia Uncommon - Foreign body - Cystic fibrosis - Bronchiopulmonary displasia Rare - Congestive heart failure - Mediastinal mass - Bronchiolitis obliterans - Trachobronchila anomalies |
|
|
|
Cause of wheezing
|
Lower respiratory tract disease
|
|
|
|
Pneumonia in neonates
- Organisms (bacteria, viral, atypical) - Treatment |
Bacterial : BGS, E. Coli, Listeria
Viral : CMV, HSV, Enterovirus Atypical : Mycoplasma hominis, ureaplasma urealyticum Treatment Ampicillin + Gentamycin |
|
|
|
Pneumonia in children 1-3 months
- Organisms (bacteria, viral, atypical) - Treatment |
Bacterial : S. Aureus, H. Influenza, S. Pneumonia, B. Pertussis
Viral : CMV, RSV, Influena, Parainfluenza Atypical : C. Trachomatis, Ureaplasma Treatment Cefuroxime Ampicillin + Gentamycin |
|
|
|
Pneumonia in children 3 months-5 years
- Organisms (bacteria, viral, atypical) - Treatment |
Bacterial : S. Pneumonia, S. Aureus, H. Influenza, GAS
Viral : RSV, Adenovirus, Influenza Atypical : M. Pneumonia, TB Treatment Ampicillin Cefuroxime |
|
|
|
Definition of bronchiolitis
|
Defined as the first episode of wheezing associated with URTI and signs of respiratory distress
|
|
|
|
Organisms in bronchiolitis
Presentation of bronchiolitis |
VIRAL
SRV Parainfluenza Influenza Rhinovirus Adenovirus Presentation (most common during the first 2 years of life) 1. Prodrome of URTI with cough and fever 2. Feeding difficulties, irritability 3. Wheezing, respiratory distress, tachypnea, tachycardia, retractions, poor air entry lasting for 5-6 days |
|
|
|
Treatment of Bronchiolitis
|
Mild distress
- supportive : oral or IV hydration + antipyretics for fever - humidified O2 (maintain O2 sat >92%) - inhaled bronchodilator (Ventolin) 0.03 cc in 3 ml NS y mask, Q20Min, and then Q1H (stop if no response) Moderate to severe distress - as above (rarely intubation and ventilation) - Ipratoprium (Atrovent) and steroids are not effective - consider rebetol (Ribavirin) in high risk groups : bronchopulmonary dysplaisa, CHD, congenital lung disease, immunodeficient Monthly RSV-Ig or palivizumab may offer protection against severe disease in high risk patients Investigations - CXR : air trapping, peribronchial thickening, atelectasis, increased linear markings - Nasopharyngeal swab : direct detection of viral antigen - WBC usually normal |
|
|
|
Indications for hospitalization for bronchiolitis
|
- Hypoxia : SaO2 <92% on initial presentation
- Persistent resting tachypnea >60 RPM and retractions after several salbutamol masks - Past history of chronic lung disease, hemodynamically significant cardiac disease, neuromuscular problem, immunocompromised - Young children <6 months old - Significant feeding problems - Social problem |
|
|
|
Coloboma
|
A coloboma (from the Greek koloboma, meaning defect,[1] and also part of the rare Cat eye syndrome) is a hole in one of the structures of the eye, such as the iris, retina, choroid or optic disc. The hole is present from birth and can be caused when a gap called the choroid fissure between two structures in the eye, which is present early in development in the uterus, fails to close up completely before a child is born. The classical description in medical literature is of a key-hole shaped defect. A coloboma can occur in one or both eyes.
|
|
|
|
Diseases screened at birth
|
'Metabolic' diseases
Congenital hypothyroidism Congenital adrenal hyperplasia Phenylcetonuria Galactosemia Sickle cell disease (at risk groups) Maple syrup urine disease Homocystinuria Biotinidase deficiency Metabolic disease should be ruled out in any newborn who becomes acutely ill after a periods of normal behaviour and development or with a family history of early infant death. |
|
|
|
Phenylketonuria
|
Incidence 1 : 10 000
Screened in all newborns Etiology : deficiency of phenylalanine hydroxylase prevents conversion of phenylalanine to tyrosine leading to build up of toxic metabolites. Mothers who have PKU may have infants with congenital abnormalities. Presentation - Baby is normal at birth then develops a musty odour, eczema, hypertonia, tremors and mental retardation. - Hypopigmentation due to lowe tyrosine Treatment - PKU screening at birth - Dietary restriction of phenylalanine starting within the first 10 days of life - Duration of dietary restriction controversial - lifelong or until end of puberty, should be resumed during pregnancy to maintain normal phenylalanine levels. |
|
|
|
Galactosemia
|
1 : 60 000
Most commonly due to deficiency of galactose-1-phosphate uridyltransferase leading to an inability to process lactose/galactose. Increased risk of sepsis. If the diagnosis is not made at birth, liver and brain damage may become irreversible. Features : neonates who ingest galactose / lactose exhibit signs of liver and renal failure, jaundice, FTT and cataracts. Treatment : - elimination of galactose from diet (i.e. dairy, breast milk) - most infants are fed a soy based diet |
|
|
|
Tanner Stages
|
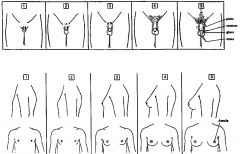
|
|
|
|
Important elements of puberty physiology
|
Activation of hypothalamo-pituitary-gonadal axis.
Adrenal production of androgens Gonadal production of hormones |
|
|
|
Normal sexual development of females
|
- Occurs between age 7-13 years (may start as early as 6 years in African-American girls)
- Usual sequence 1) Thelarche : breast budding 2) Adrenarche : axillary hair, body odour, mild acne 3) Growth spurt 4) Mearche : mean age 13 years; occurs 2 years after thelarche and indicates that growth spurt is almost complete (Tanner 4) Early puberty is common and often constitutionnal, late puberty is rare |
|
|
|
Female Tanner stages
|
1 - nada
2 - Buds + sparse labial hair 3 - Bud enlarges + hair over pubis 4 - Areola + papilla secondary mound + Coarse adult hair 5 - Areola recedes / adult size and shape + hair extends to medial thigh |
|
|
|
Normal sexual development of males
|
- Occurs between age 9-14 years (starts 2 years later than girls)
- Usual sequence 1) Testicular enlargement 2) Increase in length of penis 3) Adrenarche : axillary and facial hair, body odour, mild acne 4) growth spurt : occurs later in boys (Tanner 4) Early puberty is uncommon but late puberty is common. |
|
|
|
Male Tanner stages
|
Stage 1 : nada
Stage 2 : Scrotal enlargement + Sparse hair at base of penis Stage 3 : Increase in length of penis + Hair over pubis Stage 4 : Further increase in length of penis + coarse adult hair Stage 5 : Adult size and shape + hair extends to medial thigh |
|
|
|
Phsyiologic leukorrhea
|
Occurs 6 months prior to menarche; scant mucoid, clear to milky vaginal discharge, not associated with pruritis or foul odour.
Due to stimulation of endometrial glands by estrogen. |
|
|
|
Premature Thelarche
|
Isolated breast development in girls 6 months to 2 years.
Requires careful history and physical to ensure no other estrogen effects or other signs of puberty. May be due to increases sensitivity to estrogen. Requires observation and periodic examination. |
|
|
|
Irregular menstruation
|
Mense may be irregular in duration and length of cycle.
On average it takes 18 months to go through the first 12 periods. Birth control pills should be avoided as treatment |
|
|
|
Intubated neonate that develops pneumonia. Which germ is probably the cause?
|
Coagulase negative staphylococcus methicillin resistant.
|
|
|
|
Best way to investigate the developmental dysplasia of the hip (DDH)?
|
Ultrasound
|
|
|
|
Maneuvers on physical examination to suggest the diagnosis of developmental dysplasia of the hip.
|
Barlow
Ortolani |
|
|
|
Utility of frog leg lateral radiographs
|
Frog-leg lateral radiographs describe the hip that is flexed end externally rotated. They are performed to look for reduction if the hips are displaced or dysplastic. Plain frontal radiographs are performed more commonly, but they have no diagnostic value for DDR in infants younger than 6 months.
|
|
|
|
A 3 year old boy is brought to the ER because of a worsening cough over the past week. His temperature is 38.9 and inspiratory stridor is noted. A plain film of the neck reveals sub-glottic swelling. He is noted to have copious thick secretions and a barking cough. He has not had such events previously, and his parents deny recent contacts with sick children. The patient is in respiratory distress and is noted to be retracting his subcostal muscles to breathe. Which of the following is the next most appropriate step in management?
A) administer albuterol B) administer racemic epinephrine C) administer corticosteroids D) administer IV penicillin E) endotracheal intubation |
The patient has infectious laryngotracheitis (viral croup) which is caused by the parainfluenza virus. Symptoms are often worse at night and include a characteristic barking cough.
Fever is usllay low grade, but temperature as high as 104.0 F have been noted. Epiglottitis is in the differential but is rapidly progressive and is marked by the abrupt onset of high fever. Answer : B Racemic epinephrine by aerosol has been shown to provide symptomatic relief by vasoconstriction and reduction of local edema. This drug should be used in the moderately ill patient since it may eliminate the need for intubation. |
|
|
|
A 7 year old girl is brought to the physician because of an exanthematous rash associated with malaise and headache for 2 days. On examination, the child shows a fiery red facial rash with a characteristic "slapped cheek" pattern and pallor around the mouth. There is no fever. In immunocompromised patients, the pathogen that causes this condition may result in which of the following manifestations?
A) aplastic anemia B) Encephalitis C) Non Hodgkin's lymphoma D) Progressive multifocal leukoencephalopathy E) Symmetric polyarthritis |
Answer : A
Parvovirus B19 is the etiologic agent of this benign exanthem of childhood, which manifest with a "slapped cheeks" facial rash and little or no fever. This exanthem is referred to as fifth disease. B19 parvovirus may cause red cell aplasia in patients with AIDS or with sickle cell disease. Older people develop symmetric polyarthritis which is not seen in children. |
|
|
|
Normal Hb values by age in children
|
Newborn : 137-201
2 weeks : 130- 200 3 Months 95-145 6 Mo 6 Yrs : 105-140 |
|
|
|
Variation of MCV in children (mean corpuscular volume)
|
Lower normal limit of MCV = 70 + age until 80 fl. (adult standard)
|
|
|
|
Explanation of physiologic anemia
|
- High hemoglobin levels and reticulocyte count at birth as a result of relatively hypoxic environment in utero.
- After birth, levels start to fall due to shorter fetal RBC lifespan, decreased RBC prodution (during first 6-8 weeks of life virtually no erythropoiesis due to new O2 rich environment) and increasing blood volume secondary to growth. - Lowest levels about 100 g/L at 8-12 weeks age (earlier and more exaggerated in premature infants); levels rise spontaneously with activation of erythropoiesis. - No treatment usually required |
|
|
|
Etiology of iron deficient anemia in children
|
Dietary risk factors
- Milk's cow use - No fortified cereals after 6 months of age - Formula without iron Blood loss - Iatrogenic : repeated blood sampling - GI : peptic ulcer, IBD, chronic diarrhea - Occult bleeding / excessive bleeding - Cow's milk allergy : protein losing enteropathy + occult blood loss |
|
|
|
Mentzer index
|
The Mentzer index (MCV/RBC) allows to distinguish between iron defficient anemia and thalassemia.
Mentzer < 13 : Thalassemia Mentzer > 13 : Iron deficient anemia |
|
|
|
Prevention of iron deficient anemia in children
|
- Full term breast-fed infants : after 6 months give iron fortified cereals and iron rich foods
- Full term non breast-fed infants : use formula rich in iron + same as breast-fed - Premature infants : supply with iron from 1 month till 1 year |
|
|
|
Management of iron deficiency anemia in children
|
- Encourage diet rich in iron and limit homogenized milk to 16-20 oz a day
-Iron supplements to replenish iron stores (3 months) + Reticulocyte count increases (2-3 days, peaks day 5-7) + RBC increase (1-30 days) + Repletion of iron stores (1-3 months) + Recheck hemoglobin levels in 1 month - Poor response to oral iron therapy : / non compliance / ongoing blood loss / incorrect diagnosis / insufficient duration of therapy / high gastric pH (antacid use) |
|
|
|
Incidence of sickle cell disease
|
8% of africans carry the HbS trait
0.2% have the disease Heterozygotes are relatively malaria resistant |
|
|
|
Types of hemoglobin in sickle cell disease
|
SS
SC SD S-beta |
|
|
|
Genetic defect in sickle cell disease
|
Beta hemoglobin gene defect.
HgS : single amino acid replacement (Glutamic acid - Valine) |
|
|
|
Pathophysiology of sickle cell disease
|
Red blood cells sickle under conditions of stress : low pO2, dehydration, fever, acidosis.
Acutre intravascular sickling results in infarction of tissue (capillary occlusion and thrombosis) - spleen, lungs, bones, brain, digits. Hemolysis causes chronic, well compensated normochromic normocytic anemia ( Hb 60-90 g/L) Greates cause of mortality is infection. |
|
|
|
Presentation of sickle cell disease
|
Clinical disease presents after 5-6 months of age after fall in fetal Hb.
Anemia, fever, jaundice, splenomegaly, crisis (dactylitis is often the first presentation) Sickle cell trait : asymptomatic, may have microscopic hematuria. 2 Types of crisis : 1- Veno-occlusive crisis - due to obstruction of blood vessels by rigid, sickled cells - tissue hypoxia - cell death ; presents as fever and pain in any organ, most commonly in long bones of arms and legs, chest and abdomen, CNS (stroke), dactylitis, priapism - acute chest crisis : fever, chest pain, progressive respiratory distress, increased WBC, pulmonary infiltrates 2- Aplastic crisis : depression of erythropoiesis, generally associated with infection *parvovirus B19* 3- Acute splenic sequestration : sudden massive pooling of red cells in spleen, splenomegaly, tender spleen, acute fall in hemoglobin, shock, increased reticulocyte count. |
|
|
|
Effect of sickle cell disease on spleen
|
Splenic dysfunction usually by 5 years of age secondary to autoinfarction.
Susceptible to infection by encapsulated organisms - Meningococcus - Pneumococcus - Haemophilus type b Requires prophylactic ATB, vaccination (Men, Pneu, Hib) Immediate evaluation for any fever |
|
|
|
Long term complications of sickle cell disease
|
Increased risk of osteomyelitis (especially due to salmonella)
Growth delay Bony abnormalities (avascular nerosis of femoral head) gallstones, retinopathy, restrictive lung disease, cardiomyopathy |
|
|
|
Management of acute sickle cell disease crisis
|
1- Admit patient
2- Fluids 3- Analgesia (morphine) + ATB (ceftriaxone) + incentive spirometry 4- Straight transfusions for symptomatic, significant anemia 5- RBC exchange transfusion for impending stroke, severe chest crisis, persistent priapism 6- O2 if respiratory distress or chest crisis 7- Cultures and CBC if febrile, CXP or LP if indicated |
|
|
|
Chronic management of sickle cell disease
|
1- Early aggressive treatment of infections, prophylactic antibiotics
2- Vaccines : Pneu, Men, HBV, Hib, influenza 3- Folate supplementation 4- Hydroxyurea if recurrent crisis 5- Transcranial doppler ultrasound to assess risk of stroke 6- Chronic transfusion program 7- Annual ophtalmologic exam (after 10 years old) 8- Referral to hematology |
|
|
|
Hereditary spherocytosis
|
Red cell membrane protein abnormality, causes a sphering of RBCs which are removed by the spleen.
Genetics - Autosomal dominant - High spontaneous mutation rate Wide range of clinical severity Diagnosis : spherocytes on blood smear, osmotic fragility test Management - transfusions, splenetomy as indicated - genetic counselling |
|
|
|
G-6-PD deficiency
|
X-Linked recessive, most common enzyme deficiency worldwide
Enzyme deficient RBC unable to defend against oxidative stress (infection, drugs) --> forms Heinza bodies --> phagocytosed by splenic macrophages creating bite cells Presents with acute hemolytic anemia ( - Hemoglobinuria - Decreased haptoglobins - Increased LDH - Elevated indirect bilirubin Diagnosis : G-6-PD enzyme assay Management : supportive, hydration, transfusion, phototherapy Prevention : avoid known oxidants. G-6-PD deficiency protects against RBD paraisitism |
|
|
|
Rochester Criteria
|
Rochester criteria - developed to identify infants <60 days of age with fever (rectal >38.0) at low risk of serious bacterial infection.
- Clinically well - WBC count 5-15 x 109/L - Bands < 1.5 x 109/L - Urinalysis 10 WBC/HPF - Stool (if diarrhea) 5 WBC/HPF - Past health + Born > 37 weeks + Home with mom + No hospitalizations + No prior antibiotic use + No treated unexplained hyperbilirubinemia + No chronic disease |
|
|
|
Common organisms in Acute Otitis media in children.
|
S. Pneumoniae - 35%
H. Influenzae - 25% M. Catarrhalis 10% S. Aureus and S. pyogenes (all beta lactamase producing) Anaerobes Gram negative enterics Viral |
|
|
|
Risk factors and predisposing factors for acute otitis media in children
|
Risk factors
- bottle feeding, use of pacifier - passive smoke - crowded living conditions (day care) or sick contacts - male - family history Predisposing factors - Eustachian tube dysfunction + URTIs + Allergies, allergic rhinitis + Chronic sinusitis + Tumour (nasopharyngeal carcinoma) + Adenoid hypertrophy + Barotrauma + Inadequate tensor palati function - cleft palate + Down syndrome - Disruption of action of : + cilia of eustachian tube - Kartagener's syndrome + mucus secreting cells - Immunosuppression |
|
|
|
Pathogenesis and clinical features of acute otitis media
|
Pathogenesis
1) Obstruction of eustachian tube 2) Air absorbed in middle ear 3) Negative pressure (irritant to middle ear mucosa) 4) Edema of mucosa with exudate / effusion 5) Infection of exudate from nasopharyngeal secretions Clinical features - AOM triad (otalgia, fever, conductive hearing loss) - Rarely tinnitus, vertigo and facial nerve palsy - Otorrhea if tympanic membrane perforated - Pain over mastoid - Infants/toddlers + ear-tugging + hearing loss, balance disturbances + irritable, poor sleeping + vomiting and diarrhea + anorexia Otoscopy - Hyperemia - Bulging - Loss of landmarkds : handle and short process of malleus not visible - Reduced tympanic mobiltiy |
|
|
|
Triad of acute otitis media
|
Fever
Otalgia Conductive hearing loss |
|
|
|
Treatment of acute otitis media
|
Antibiotic treatment hastens resolution - 10 day course
1) First line - Amoxicillin 40mg/kg/day divided into two doses - if penicillin alergic : marolide (clarithromycin or azithromycin), Bactrim 2) Second line (for amoxicillin failures) - Double dose of amoxicillin (80mg/kg/day), or Augmentin - Cephalosporins : cefuroxime, ceftriaxone, cefaclor, cefixime AOM deemed unresponsive if clinical signs / symptoms and otoscopic findings persist beyond 48 hours of antibiotic treatment. Symptomatic therapy : - Antipyretics - Analgesics - Decongestants (may relieve nasal confestion but does not treat AOM) |
|
|
|
Prevention of acute otitis media
|
Parent education about risk factors
Antibiotic prophylaxis - amoxicillin or macrolide show therapeutic at half dose Pneumococcal and influenza vaccine Surgery - choice of surgical therapy for recurrent AOM depends on whether local factors (eustachian tube dysfunction) are responsible (use ventilation tubes), or regional disease factors (tonsillitis, adenoid hypertrophy, sinusitis) are responsible. |
|
|
|
Complications of AOM
|
Otologic
- TM perforation - Chronic suppurative OM - Ossicular necrosis - Cholesteatoma - Persistent effusion CNS - Meningitis - Brain abscess - Facial nerve paralysis Other - Mastoiditis - Labyrinthitis - Sigmoid sinus thrombophlebitis |
|
|
|
Indications for myringotomy and tympanostome tube in recurrent AOM and OME
|
Tubes are more commonly inserted for OME, rarely for AOM.
- Persistent effusion > 3months (OME) - Lack of response to > 3 months of ATB therapy - Persistent effusion for > 3 months after episode of AOM - Recurrent episodes of AOM (> 7 episodes in 6 months) - Bilateral conductive hearing loss of > 20 dB - Chronic retraction of the tympanic membrane or pars flaccida (OME) - Bilateral OME lasting > 4 to 6 mos |
|
|
|
Complications of tympanostomy tubes
|
Early
- Extrusion - Blockage - Persistent otorrhea Late - Myringosclerosis - Persistent TM perforation - Cholesteatoma |
|
|
|
Definition of otitis media with effusion
|
Presence of fluid in the middle ear without signs or symptoms of ear infection.
Middle ear effusions have been shown to persist following an episode of AOM for 1 mo in 40% of children, 2 mo in 20% and 3+ mo in 10%. Same risk factors as AOM. |
|
|
|
Otoscopy of tempanic membrane in otitis media with effusion
|
Discolouration
Meniscus fluid level Air bubbles Retraction pockets Immobility |
|
|
|
Complications of otitis media with effusion
|
Hearing loss, speech delay, learning problems in young children
Chronic mastoiditis Ossicular erosion Cholesteatoma Retraction of tympanic membrane, atelectasis, ossicular fixation |
|
|
|
Clinical features of adenoid hypertrophy
|
Nasal obstruction
- adenoid facies (open mouth, flat midface, dark circles under eyes) - hypernasal voice - history of snoring - long term mouth breather, minimal air escape through nose Choanal obstruction - chronic sinusitis - obstructive sleep apnea Chronic inflammation - nasal discharge - cervical adenopathy Size peaks at age 5 and resolves by 12 to 18 years of age Increase in size with repeated URTIs and allergies |
|
|
|
Types of tonsils
|
Pharyngeal tonsil (adenoid)
Tubal tonsils (auditory) x 2 Palatine tonsils Lingual Tonsils x 2 |
|
|
|
Diagnosis of adenoid hypertrophy
|
- Enlarged adenoids on direct / indirect nasopharyngeal exam
- Enlarged adenoid shadow on lateral soft tissue X-Ray - Lateral view of the nasopharynx may show a large pad of adenoidal tissue |
|
|
|
Complications of adenoid hypertrophy
|
Eustachian tube obstruction leading to serous otitis media
Interference with nasal breathing, necessitating mouth-breathing Malocclusion Sleep apnea Orofacial developmental abnormalities |
|
|
|
Indications for adenoidectomy
|
Chronic upper airway obstruction with sleep disturbance / apnea or cor pulmonale
Chronic nasopharyngitis resistant to medical treatment Chronic serous OM and chronic suppurative OM Recurrent AOM resistant to antibiotics Suspicion of nasopharyngeal malignancy Orofacial developmental abnormalities |
|
|
|
Common organisms in acute tonsillitis
|
GABHS
Group G streptococci S. Pneumoniae S. Aureus H. Influenzae M. Catarrhalis EBV |
|
|
|
Clinical features of acute tonsillitis
|
Symptoms
- sore throat - dysphagia, odynophagia, trismus - malaise, fever - otalgia (referred) Signs - tender cervical lymphadenopathy especillay submandibular, jugulodigastric - tonsils enlarged, inflammation +/- eudates - strawberry tongue, scarletinifrom rash (scarlet fever) - palatal petechiase (infectious mononucleosis) |
|
|
|
Differential diagnosis for sore throat
|
Strep Pharyngitis
Tonsillitis EBV Viral pharyngitis Peritonsollar abscess Foreign body / trauma Leukemia Hodgkin's disease |
|
|
|
Trismus
|
Motor disturbance of the trigeminal nerve, leading to spasm of masticatory musles, with difficulty in opening the mouth (lockjaw).
|
|
|
|
Treatment of acute tonsillitis
|
Bed rest, soft diet, ample fluid intake
Gargle with warm saline solution Analgesics and antipyretics Antibiotics - Only after appropriate swab for C&S - 1st line penicillin or amoxicillin (macrolide if PCN allergy) x 10 days Rheumatic fever risk emerges approximately 9 days after the onset of symptoms : antibiotics are utilized mainly to avoid this serious sequela and to provide earlier symptomatic relief. No evidence for the role of antibiotics in the avoidance of post-streptococcal GN. |
|
|
|
Acute tonsillitis complications
|
Deep neck space infection
Abscess : peritonsillar, intratonsillar Sepsis Glomerulonephritis Rare : - Rheumatic heart disease - Arthritis - Scarlet fever - Quinsy (Peritonsillar abscess) |
|
|
|
What is a peritonsillar abscess?
|
Cellulitis of spae behind tonsillar capsule extending onto soft palate leading to abscess.
Also referred to as Quinsy Same bacterial organisms as acute tonsillitis. |
|
|
|
Quinsy triad
|
Trismus
Uvular deviation Hot potato voice |
|
|
|
Clinical features of acute tonsillitis
|
Fever and dehydration
Sore throat, dysphagia and odynophagia Extensive peritonsillar swelling but tonsil may appear normal Edema of soft palate Uvular deviation Involvement of motor branch of CN V; can lead to increased salivation and trismus Dysphonia with "hot potato" voice (edema --> failure to elevate palate) secondary to CN X involvement Unilateral referred otalgia Cervical lymphadenitis |
|
|
|
Complications of Quinsy
|
Aspiration pneumonia 2nd to spontaneous rupture of abscess
Airway obstruction Lateral dissection into parapharyngeal and / or carotid space Bacteremia |
|
|
|
Tonsillectomy indications (absolute and relative)
|
Absolute indications
- Acute airway obstruction +/- cor pulmonale - Suspected malingnancy, especially if unilateral tonsillar hypertrophy - Acute hemorrhage Relative indications - age 1 to 4 years : tonsillar hypertrophy leading to : + sleep apnea (cor pulmonale) + chronic nasal obstruction or mouth breathing + difficulty swallowing -- FTT + speech abnormalities + severe orofacial / dental abnormalities - school age : chronic recurrent tonsillitis if 4-7 episodes in 1 year - any complication of tonsillitis |
|
|
|
Signs of croup
|
Stridor
Subglottic swelling Seal bark cough |
|
|
|
Common organisms in croup
(tracheolaryngobronchitis) |
Parainfluenza virus
Influenza virus A & B RSV |
|
|
|
Sign of croup on neck x-ray
|
Steeple sign
|
|
|
|
Treatment of tracheolaryngobronchitis
|
1- Racemic epinephrine
2- Systemic corticosteroids 3- Adequate hydration 4- Close observation for 3 to 4 hours 5- Intubation if severe 6- Hospitalize if poor response to steroids after 4 hours and persistent stridor at rest. |
|
|
|
CSF findings of meningitis
|

|
|
|
|
Viral vs Bacterial pharyngitis
|

|
|
|
|
When to initiate antibiotic therapy for acute otitis media?
|
In children older > 6 years, it is appropriate to consider a "wait and see" prescription with instructions for re-evaluation if the child has not improved significantly within 72 hours.
In children < 2 years with bilateral AOM or AOM with otorrhea, antibiotics are recommended by a recent meta analysis. Recommendations Treat immediately with antibiotics when : - Age < 6 years - Ill appearance - Recurrent AOM - Suspicion of another concurrent bacterial illness - Recent treatment with antibiotics (within last 7 days) - Perforated TM (including tympanostomy tubes) - Immunocompromised - Craniofacial abnormalities - Poor access to medical care |
|
|
|
Common organisms in meningitis (children)
|
0-3 months : Strep B, E.Coli, L. monocytogenes, GNB, viral
3 mo - 3 yrs : S. Pneumoniae, N. Meningitidis, H. Influenzae, TB, viral 3-18 years : S. Pneumoniae, N. Meningitids, H. Influenzae, viral (enterovirus, adenovirus, HSV) |
|
|
|
Signs of meningismus in children
|
Bluging Fontannelle
Brudzinski Kernig Nucchal rigidity Opisthotonos |
|
|
|
Brudzsinki's sign
|
Reflex flexion of hips and knees upon active flexion of the neck
|
|
|
|
Kernig's sign
|
Reflex contraction and pain in hamstrings upon extension of leg that is flexed at the hip.
|
|
|
|
Opisthotonos
|
Spasm in which head and heels are bent backward and body bowed forward.
|
|
|
|
Normal opening pressure in lumbar puncture
|
< 160 mm H2O
Flexed lateral decubitus position : 100-280 mm H2O |
|
|
|
Acute and chronic complications of meningitis
|
Acute complications
- SIADH - Subdural effusion / empyema - Brain abscess, disseminated infection - Shock / DIC Chronic complications - Hearing loss - Intellectual disability, learning disorders - Neurological deficits, seizure disorder - Hydrocephalus |
|
|
|
Management of meningitis in children
|
- Isolation with appropriate infection control procedures until 24hr after culture-sensitive antibiotic therapy.
- Bacterial : empiric antibiotics + Neonatal : Ampicillin + gentamycin + 6 weeks - 3 Mo : Ampicillin + cefotaxim with vancomycin if s. aureus + > 3 Mo : Ceftriaxone + Vancomycin - Monitor : glucose, acid-base and volume status - Anticonvulsants may be needed to treat seizures - Prophylaxis + Hib vaccine + Men C vaccine + Pneu C vaccine - Antibiotic prophylaxis for close contacts - Report to public health |
|
|
|
McIsaac criteria
|
GAS pharyngitis
- Fever > 38 - Lymphadenopathy - anterior, tender, cervical - Age 3-14 - No cough - Erythematous, exudative tonsils Score 0-1 : no culture, no antibiotic Score 2-3 : culture, treat if positive Score 4 : antitiobitics |
|
|
|
Management of Streptococcal pharyngitis
|
- > 2 years old, culture before treatment or do rapid Strep antigen test (Sn 70-90%), culture if negative
- Symptomatic + if 1 symptoms : no culture, no ATB + if > 1 symptoms, culture then ATB - Penicillin V or amoxil 40mg/kg/day PO divided bid x 10 days - Erythromycin 40mg/kg/day PO divided tid x 10 days if allergic to penicillin - Acetaminophen for discomfort + Can prevent rheumatic fever if treated within 9-10 days + ATB do not alter the risk of post-streptococcal glomerulonephritis + Tonsillectomy for proven, recurrent streptococall tonsillitis |
|
|
|
Complications of streptococcal pharyngitis
|
- If untreated, can lead to :
+ suppurative complications : AOM, sinusitis, cervical adenitis, pneumonia, mastoiditis + direct extension : retropharyngeal / pertitonsillar abscess + scarlet fever, rheumatic fever + hematogenous spread : bone/joint infection, meningitis, SBE - Acute glomerulonephritis - Invasive GAS disease : illness associated with isolation of GAS from normally sterile sites. -Other illnesses caused by strep : impetigo, cellulitis, bacteremia, vaginitis, toxic shock syndrome |
|
|
|
Treatment of invasive Group A Streptococcal infection
|
Admit
Clindamycin 40mg/kg divided tid or qid + IV penicillin 250 000-400 000 U/kg/day |
|
|
|
Scarlet fever
|
Erythrogenic strain of Group A Streptococcus
= Acute onset of fever, sore throat, strawberry tongue 4 S and 4 P Sore throat Strawberry tongue Sandpaper rash Perioral Sparing Non Pruritic Non Painful Peeling 24-48 hours after pharyngitis, rash begins in the groin, axillae, neck, antecubital fossa Within 24 hours, sandpaper rash becomes generalized with perioral sparing, non pruritic, non painful. Rash fades after 3-4 days, may be followed by peeling. Treatment : same as GAS pharyngitis |
|
|
|
Jones Criteria (Revised)
|
Rheumatic Fever requires :
+ 2 Major + 1 Major and 2 minor plus evidence of preceding strep infection (scarlet, culture, strep test, ASLO) Major criteria "SPACE" - S : subcutaneous nodules, pea-sized, firm, non tender nodules typically on extensor surfaces - P : Pancarditis involving perdicardium, myocardium, endocardium - A : Arthritis (migratory) - C : Chorea (sydenham) - Erythema marginatum : begins as pink macules on trunk with central blanching, non pruritic Minor criteria - Previous history of rheumatic fever or rheumatic heart disease - Polyarthralgia - Fever - Elevated ESR or C-reactive protein or leukocytosis - Prolonged PR interval |
|
|
|
Treatment of rheumatic fever
|
Acute
- Penicillin or erythromycin x 10d - ASA for arthritis - Prednisone for severe carditis Secondary prophylaxis with daily penicillin or erythromycin; course depends on: - without carditis : 5 years or until 21 years old, whichever is longer - with carditis but no residual heart disease : 10 years or longer - carditis and residual heart disease : at least 10 years since last episode, sometimes life long |
|
|
|
Complications of rheumatic fever
|
Acute : myocarditis, conduction system abnormalities, valvulitis, pericaditis
Chronic : - Rheumatic heart disease - Mitral and/or aortic insuffisiency/ stenosis - Increased risk of infectious endocarditis - Onset of symptoms usually after 10-20 year latency from acute carditis of rheumatic fever |
|
|
|
Clinical features of infectious mononucleosis
|
EBV infection
- Prodrome : 2-3 days of malaise, anorexia - Infants and young children : often asymptomatic or mild malaise - Older children and young adults : typical mononucleosis syndrome + Fever, tonsillar exudate, generalized lymphadenopathy, pharyngitis + +/- Hepatosplenomgaly + +/- Rash (rash more frequent with patients treated with amoxicillin/ampicillin + Any -itis (arthritis, hepatitis, nephritis, myocarditis) + Chronic fatigue - Resolves over 2-3 weeks although fatigue may persist for several months - Administration of amoxicillin results in rash in >90% of cases |
|
|
|
Complications of infectious mononucleosis
|
Aseptic meningitis
Encephalitis Guillain-Barré Splenic rupture Agranulocytosis Myocarditis |
|
|
|
Diagnosis of infectious mononucleosis
|
MonoTest : heterophile antibody test (85% sensitivity in adults and older children, only 50% sensitive <4 yrs of age
+ False positive result with HIV, SLE, Lymphoma, Rubella, Parvovirus EBV titres CBC + differential : atypical lymphocytes, lymphocytosis, Downey cells +/- anemia/thrombocytopenia |
|
|
|
Downey Cells
|
Indicate EBV infection
|
|
|
|
Treatment of infectious mononucleosis
|
- Throat culture to r/o streptococcal pharyngitis
- Supportive care (bed rest, fluids, saline gargles for sore throat, tylenol) - If airway obstructed secondary to node and/or tonsillar enlargement, admit to hospital, STEROIDS - Patients with splenic enlargement often not apparent clinically so all patients should avoid contact sports for 6-8 weeks - Acyclovir not usedul |
|
|
|
Pertussis
|
B. Pertussis, whooping cough, "100 day cough"
Incubation : 6-20 days, infectivity : 1 week before paroxysms to 3 weeks after. Highly contagious via air droplets released during intense coughing. Clinical Presentation : - Prodromal catarrhal stage + 1-2 weeks, most contagious + coryza, mild cough - Paroxysmal stage + 2-4 weeks + paroxysms of cough, sometimes followed by inspiratory whoop + infants may present with apnea + vomiting with coughing spells + pressure effect : subconjunctival hemorrhage, rectal prolapse, hernia, epistaxis - Convalescent stage + 1-2 weeks, noninfectious + occasional paroxysms of cough, but decreased frequency and severity, lasts up to 6 months |
|
|
|
Pertussis diagnosis
|
Clinical : URTI symptoms followed by paroxysms of cough in an afebrile child.
PCR of nasopharyngeal swab or aspirate. |
|
|
|
Complications of pertussis
|
AOM
URTIs Pneumonia, atelectasis, pneumomediastinum, pneumothorax, empyema Neurological : seizures, encephalopathy, intracranial hemorrhage. |
|
|
|
Treatment of Pertussis
|
- Supportive care
- Hospitalize if paroxysms of cough are associated with cyanosis and/or apnea (O2) - Erythromycin 40 mg/kg/day x 10 days started within 3 weeks after onset of cough + isolate until 5 days of treatment + treatment will decrease infectivity but not change course of illness + shortens period of communicability - Antibiotic prophylaxis : erythromycin for all household contacts - Prevention : acellular pertussis vaccine (Pentacel) in infants and children, and pertussis booster (Adacel) in adolescents and children. |
|
|
|
Varicella
|
VZV
Incubation : 10-21 days, infectivity :1-2 days pre-rash until vesicles have crusted over Transmission rate is 86% in household contacts, via respiratory secretions and vesicular fluid Primary infection with virus usually results in life-long immunity : >95% of young adults with varicella are immune. Clinical Presentation - 1-3 day prodrome : fever and respiratory symptoms - Characteristic polymorphous rash + Very pruritic + Crops of red macules which quickly become vesicles surrounded by erythema + "dewdrop" on erythematous base + Vesicles burst and lesions crust over + On trunk, scalp, face, conjunctivae, vagina, oral mucosa, palms and soles + New crops usually stop forming after 5-7 days |
|
|
|
Complications of varicella
|
- Secondary bacterial infection
+ infection with staph, GAS + presents as impetigo, abscesses, cellulitis, necrotizing fasciitis, sepsis - Cerebellar ataxia, pneumonia, hepatitis, encephalitis - Immunocompromised patients : varicella may be life threatening - Virus latent in sensory ganglia and reappears as herpes zoster in 68/100 000 individuals |
|
|
|
Rx used to delay peripheral precocious puberty
|
Ketoconazole
5 alpha reductase inhibitors (finasteride) Steroid receptor blockers (spironolactone) Aromatase blockers (Anastrozole, testolactone) |
|
|
|
Causes of short stature
|
Decreased Growth Velocity
- Primordial (Ht, Wt, HC are affected) * chromosomal (turner, down, dysmorphism) * skeletal dysplasia * Intra uterine growth restriction - Endocrine (Ht affected more then weight) - short and fat * GH deficiency * Hypothyroidism * Hypocorticism * Hypopotuitarism - Chronic disease * Cyanotic congenital heart disease * Celiac disease, IBD, CF * Chronic infections * CRF - Psychosocial neglect Normal Growth velocity - Constitutional growth delay (delayed bone age) - Familial (Normal bone age) |
|
|
|
Formula to predict adult height
|
Boy = ((Father height + mother height) + 13 ) / 2
Girl = ((Father height + mother height) - 13 ) / 2 centimeters :) |
|
|
|
Diagnosis of short stature
|
Stature < 3rd percentile
Height crosses 2 major percentiles Growth velocity < 25th percentile |
|
|
|
Investigations for short stature
|
Bone age
Karyotype in girls to r/o Turner Other tests according to history |
|
|
|
Role of GH in growth
|
GH is important for chondrocyte proliferation and IGF-1 release.
IGF-1 acts at long bones, liver, negative feedback |
|
|
|
Etiology in GH deficiency
|
Congenital GH deficiency
- idiopathic - embryologic CNS malformation - perinatal asphyxia - rare mutations Acquired GH deficiency Tumours (e.g. craniopharyngioma) Trauma Cranial infection Irradiation Hypoglycaemia |
|
|
|
Clinical presentation of GH deficiency
|
Infantile features and fat distribution (short and chubby)
Delayed puberty Hypoglycemia High pitched voices |
|
|
|
Investigations for GH deficiency
|
Stimulation testing only performed when :
- height < 3rd percentile - decreased growth velocity - midline craniofacial anomalies - episodes of hypoglycemia - delayed bone age, puberty |
|
|
|
Causes of physiologic increase of GH
|
Clonidine
Arginine Insuline / Hypoglycemia Dopamine Propranolol |
|
|
|
Value to say that GH stimulation is positive
|
If GH fails to raise > 8-10 ng /ml post stimulation
|
|
|
|
Causes of acute diarrhea in children
|
Same as adults.
Do not forget Hirschsprung's disease |
|
|
|
Normal stool volume in children
|
Infants : 5-10 g/kg
Children : 200 g/day |
|
|
|
Most common cause of diarrhea in children
|
Viral infection
1. Rotavirus (especially prevalent during winter months) 2. Astrovirus 3. Norwalk Virus Often associated with URTIs Resolves in 3-7 days Slight fever, vomiting, vague abdominal pain |
|
|
|
Indications for medical evaluation of acute diarrhea
|
Age < 6 months
Fever Visible blood in stool Frequent, substantial volume of diarrhea Signs of dehydration Change in mental status |
|
|
|
Management of diarrhea in children
|
Prevention and treatment of dehydration is most important
- Oral rehydration therapy with frequent small volumes of pediatric rehydration solutions, IV may be required - Early refeeding advisable, start with small amounts of easily digested carbohydrates, postpone dairy and fibrous vegetables Antibiotic therapy when indicated Antidiarrheal medications not indicated Notify public health authorities if appropriate |
|
|
|
Definition of chronic diarrhea in children
|
Three or more loose, watery stools per day lasting > 14 days
|
|
|
|
Investigations for chronic diarrhea
|
Perform serial heights, weights, growth percentiles
If child is growing well and thriving, minimal workup is required. Investigations depending on suspected diagnosis : - Stool : consistency, pH, reducing substances, microscopy, occult blood, O&P, C&S, C. Difficile toxin, 3 day fecal fat - Urinalysis - CBC, differential, ESR, smear electrolytes, total protein, albumin - Absorptive and nutritional status : albumin, carotene, Ca, PO4, Mg, Fe, Ferritin, Folate, fat soluble vitamins, PTT, INR - Sweat chloride, TSH, Urine vanillyl mandellic acid and homovallinic acid, HIV test, Lead - CXR, upper GI series and follow through |
|
|
|
Common causes of chronic diarrhea according to age
|
0-3 Months : GI infection, Disaccharidase infection, Cow's milk intolerance, Cystic fibrosis
3 MO- 3 yrs : Toddler's diarrhea, GI infection, Celiac disease 3 yrs- 18 yrs : GI infection, Celiac disease, IBD Uncommon causes : constipation with overflow diarrhea, drug induced, UTI, Short bowel syndrome |
|
|
|
Causes of chronic diarrhea without failure to thrive
|
Infectious
- Bacterial : Campylobacter, Salmonella - ATB induced : C. difficile colitis - Parasitic : Giardia lamblia - Post-infectious : secondary lactase deficiency Toddler's diarrhea Lactase deficiency Irritable bowel syndrome |
|
|
|
Definition and presentation of toddler's diarrhea
|
Epidemiology
- most common cause of chronic diarrhea during infancy - onset between 6-36 months of age, cases spontaneously between 2-4 years Clinical presentation - Diagnosis of exclusion in thriving child (no weight loss / FTT, no fluid or electrolye abnormalities) - Diet history : too much juice overwhelms small bowel resulting in disaccharide malabsorption - Stool may contain undigested food particles, 4-6 bowel movements per day - Excoriated diaper rash |
|
|
|
Management of toddler's diarrhea
|
Reassurance (self limiting)
Four F's - adequate fibre - normal fluid intake - 35-40% Fat - discourage excess Fruit juice |
|
|
|
Definition and presentation of lactase deficiency
|
Clinical features
- chronic, watery diarrhea - abdominal pain, bloating associated with dairy intake Primary lactose intolerance : crampy abdominal pain with loose stool (usually of east asian and african descent) Secondary lactose intolerance : older infant, persistent diarrhea (post viral / bacterial, infections, celiac disease, or IBD) |
|
|
|
Diagnosis of lactase deficiency
|
Trial of lactose free diet
Watery stool, acid pH, positive reducing sugars Positive breath hydrogen test if >6 years |
|
|
|
Management of lactase deficiency
|
Lactose free diet, soy formula
Lactase containing tablets/capsules/drops - Lacteeze - Lactaid |
|
|
|
Which test should be ordered when dosing anti transglutaminase antibodies?
|
IgA levels because celiac disease can be associated with IgA deficiency.
|
|
|
|
Non GI manifestations of celiac disease
|
Dermatitis herpetiformis
Dental enamel hypoplasia Osteopenia/Osteoporosis Short stature Delayed puberty |
|
|
|
Grains to avoid with celiac disease
|
Wheat
Barley Rye Oat |
|
|
|
Common causes of chronic diarrhea with failure to thrive
|
Intestinal causes
- Celiac disease - Milk protein allerhy - IBD - Specific enzyme deficiencies - Liver diease - Biliary atresia - Alpha beta lipoproteinemia - Short gut toxic or immunologic reaction - Blind loop syndrome - Giardiasis Pancreatic insufficiency - Cystic fibrosis - Schwachmann-Diamond syndrome Other - Diets rich in sorbitol, fructose - Metabolic / endocrine : thyrotoxicosis, Addison's, Galactosemia - Immune defects : IgA deficiency, AIDS, hypogammaglobulinemia, severe combined immunodeficiency - Neoplastic : pheochromocytoma, lymphoma of small bowell, carcinoid tumours, secretory tumours - Food allergy - Laxative abuse |
|
|
|
What is milk protein allergy
|
Immune mediated mucosal injury.
Up to 50% of children intolerant to cow's milk may be intolerant to soy protein as well. Often history of atopy |
|
|
|
Presentation of milk protein allergy
|
2 Scenarios :
- enterocolitis : vomiting, diarrhea, anemia, hematochezia - enteropathy : chronic diarrhea, hypoalbuminemia |
|
|
|
Schwachman-Diamond Syndrome
|
Pancreatic insufficiency
Cyclic neutropenia Anemia Accompanied with skeletal abrnomalities (metaphyseal dysostosis leading to short stature) |
|
|
|
How to differentiate pancreatic insufficiency from cystic fibrosis
|
Schwachmann Diamond syndrome is distinguished from cystic fibrosis by normal sweat chloride test, characteristic metaphyseal lesions, fatty pancreas on CT.
|
|
|
|
Definition of constipation in children
|
Decreased stool frequency
< 3 stools / week |
|
|
|
Causes of constipation in children
|
Functional constipation
Hirschsprung's disease Endocrine : hypothyroidism, diabetes mellitus, hypercalcemia Neurologic : spina bifida Anatomic : bowel obstruction, anus (imperforate, atresia, stenosis) Drugs : lead, chemotherapy, opioids |
|
|
|
Functional constipation in children
|
99% of cases of constipation
Lack of fibre in diet or change in diet, poor fluid intake Infants: when introducing cow's milk after breast milk because increased fat and solute amounts, lower water content Toddlers / older children : can occur during toilet training, or due to pain on defecation |
|
|
|
Complications of constipation in children
|
Pain retention cycle : anal fissures and pain - withhold passing stool - chronic dilatation and overflow incontinence
|
|
|
|
What is meconium
|
econium is the earliest stools of an infant. Unlike later feces, meconium is composed of materials ingested during the time the infant spends in the uterus: intestinal epithelial cells, lanugo, mucus, amniotic fluid, bile, and water.
|
|
|
|
Meconium ileus
|
The meconium sometimes becomes thickened and congested in the ileum, a condition known as meconium ileus. Meconium ileus is often the first sign of cystic fibrosis.[5] In cystic fibrosis, the meconium can form a bituminous black-green mechanical obstruction in a segment of the ileum.
|
|
|
|
Management of constipation in children
|
Adequate fluid intake (if <6 months, 150 ml/kg/day
Adequate dietary fibre, mineral oil, gentle laxatives occasionally Appropriate toilet training technique |
|
|
|
What is Hirschsprung's disease?
|
Congenital aganglionic megacolon.
Failure of normal innervation of the distal colon by the ganglion cells of the myenteric plexus. Colon remains contracted and impairs faecal movement. |
|
|
|
Clinical features of Hirschsprung's disease
|
Typically only rectosigmoid involvement but may extend to entire colon
No meconium within first 24 hours Palpable stool on abdominal exam with empty rectum on DRE Intermittent diarrhea, BM only with rectal stimulation Constipation, abdominal distention, vomiting, FTT |
|
|
|
Complications of Hirschsprung's disease
|
Enterocolitis
Toxic megacolon and perforation |
|
|
|
Diagnosis of Hirschsprung's disease
|
Abdominal X-Ray (cannot see gas in rectum)
Barium enema (proximal dilatation due to functional obstruction) Manometric studies : shows failure of anal sphincter relaxation Rectal biopsy : definitive diagnosis (absent ganglion cells) |
|
|
|
Treatment of hirschsprung's disease
|
Non surgical if short segment
Surgical : colostomy and re-anastomosis |
|
|
|
Differential diagnosis of acute abdominal pain
|
Gastrointestinal
- Gastroenteritis - Incarcerated hernia, volvulus, intussusception - Appendicitis - Malrotation - Mesenteric adenitis - Cholecystitis - Meckel's diverticulum Other systems - UTI - Henoch-Schonlein Purpura - Sickle cell crisis - Penumonia - DKA - Nephrolithiasis - Gynecological (ectopic pregnancy, PID, endometriosis, menstruation) |
|
|
|
Classic triad of intussusception
|
Abdominal pain
Palpable mass Red current jelly stools |
|
|
|
What is intussusception?
|
Telescoping of segment of bowel into distal segment causing ischemia and necrosis
|
|
|
|
Usual site of intussusception?
|
Ileocecal junction
Jejunum in children with GJ tubes |
|
|
|
Clinical features of intussusception?
|
Classic triad :
- palpable abdominal mass - abdominal pain - red current jelly stools Sudden onset pf recurrent, paroxysmal, severe periumbilical pain with pain-free intervals Later vomiting and rectal bleeding Shock and dehydration 50% between 3-12 months, 75% before 2 years of age 90% idiopathic |
|
|
|
Diagnosis and treatment of intussusception
|
U/S
Air enema diagnostic Reduction under hydrostatic pressure Surgery rarely needed |
|
|
|
Differential diagnosis of abdominal mass in children
|

|
|
|
|
Definition of chronic abdominal pain in children
|
3 episodes of pain severe enough to affect activities, occurring in a child > 3 years of age over a period of 3 months.
|
|
|
|
Causes of chronic abdominal pain in children
|
Organic (<10%)
- GI : constipation, infectious, IBD, esophagitis, PUD, lactose intolerance, anatomic anomalis, pancreatic, hepatobiliary - GU : recurrent UTIs, nephrolithiasis, chronic PID, mittelschmerx - Neoplastic Functional / Recurrent abdominal pain (90%) |
|
|
|
Mittelschmerz
|
Mittelschmerz is characterized by lower abdominal and pelvic pain that occurs roughly midway through a woman's menstrual cycle. The pain can appear suddenly and usually subsides within hours, although it may sometimes last two or three days.
|
|
|
|
What is functional / recurrent abdominal pain?
|
School age, peak 8-10 years
Prevalence : 10% of school children, F>M Characteristics : - Vague, crampy periumbilical or epigastric pain, vivid imagery to describe pain, clustering of episodes - Seldom awakens child from sleep - Aggravated by exercice, alleviated by rest Psychological factors related to onset and / or maintenance of pain, school avoidance Psychiatric comorbidity Diagnosis by exclusion of organic disorders |
|
|
|
Treatment of functional abdominal pain
|
Continue to attend school
Manage any emotional or family problems, counselling Trial of high fibre diet, trial of lactose free diet Reassurance |
|
|
|
Prognosis of functional abdominal pain in children
|
Pain resolves ni 30%-50% of kids within 2-6 weeks of diagnosis.
30-50% of kids with RAP have function pain as adults (IBS). |
|
|
|
Red flags for organic etiology of chronic abdominal pain
|
Age < 5 years old
Fever Localizes pain away from midline Anemia Rectal bleeding Rash Pain awakens child at night Travel history Prominent vomiting, diarrhea Weight loss or FTT Joint pain |
|
|
|
Causes of upper GI bleed in children
|
Same as adults
|
|
|
|
Causes of lower GI bleed in children
|
Acute
- infectious - bacterial, parasitic, antibiotic induces (C. Difficile) - necrotizing enterocolitis in preterm infants - anatomic * malrotation / volvulus / intussusception * Meckel's diverticulum * anal fissure, hemorroids - Vascular / hematological * Henoch-Schonlein Purpura * Hemolytic uremic syndrome * Coagulopathy Chronic - anal fissures (most common) - colitis - IBD - allergic - structural * polyps * neoplasms - coagulopathy |
|
|
|
Investigations for lower GI bleeding children
|
Hemodynamic status, evidence of FTT, fever
Anal and rectal exam - tags, fissures, anal fistulas, polyps - foreign body - blood - stool appearance NG aspirate Stool cultures + C. difficile toxins Urinalysis and microscopy CBC, smear, platelets, ESR, electrolytes, urea, creatinine, INR, PTT, albumin, iron studies, amoeba titers Abdominal X-Ray to r/o obstruction |
|
|
|
Usual presentation of ureterovesical reflux
|
Recurrent UTI
|
|
|
|
DiGeorge syndrome
|
Fetal malformations of the third and fourth branchial arches. It causes failure of development of both the parathyroid glands and the thymus. An abnormal face with a small jaw may be noted at birth.
The absence of the parathyroid glands causes hypocalcemic convulsions and tetany in infancy. Aplasia or near aplasia of the thymus predisposes to infections. Circulating T cells may be very low to absent. The presence of even a small amount of functioning thymic tissue may allow these children to outgrow their immune deficiency. |
|
|
|
If child abuse is suspected, what are important aspects to look for?
|
Retinal hemorrhage (fundoscopic exam) --> First thing to verify
Rib Fractures (CXR) Head injury (CT-Scan) |
|
|
|
Wiskot aldrich syndrome
|
X-linked condition
Characterized by : - Thrombocytopenia (with characteristically small platelets) - Lymphopenia with depressed cellular immunity Typically accompanied by : - High serum IgE - Low serum IgM Atopic eczema is very common and may be the presenting complaint in these patients. These patients are very prone to develop non Hodgkin's lymphoma. |
|
|
|
Neoplasia association with these conditions :
- Wiskot aldrich - Large congenital nevi - Xeroderma pigmentosa - Basal cell nevus syndrome |
Wiskot Aldrich : Non hodgkin's lymphoma
Large congenital nevi : Melanoma Xeroderma pigmentosa : Basal cell carcinoma and squamous cell carcinoma Basal cell nevus syndrome : squamous cell carcinoma |
|
|
|
What is the most important determinant of the neuro-developmental outcome of any very low birth weight infant?
|
Gestation length.
|
|
|
|
Serum iron levels in lead intoxication
|
High serum iron levels because lead inhibits ferrochelatase, thus inhibiting the production of hemoglobin,
|
|
|
|
What does valine to glutamate substitution indicate?
|
Sickle cell anemia
The amino acid substitution leads to polymerization of hemoglobin in low oxygen states, causing sickling and hemolysis and the peripheral blood smear shows characteristic sickling of red cells. The cells will also be hypochromic secondary to the low iron content. |
|
|
|
Nocturnal bouts of pain in a child that are promptly relieved by aspirin.
Which disease should be suspected? |
Osetoid osteoma
XR : radiolucent nidus surrounded by a wide rim of osteosclerosis in a typical location. Typically this benign tumour is located in the metaphyseal cortex of long bones (femur, tibia) Treated with surgical resection. |
|
|
|
Osetoid osteoma
|
Nocturnal bouts of pain in a child that are promptly relieved by aspirin.
|
|
|
|
Enchondroma
|
Benign cartilaginous tumor that most frequently develops in the medullary cavity og phalangeal, metacarpal, and metatarsal bones.
X-Ray : osteolytic area in the diaphysis surrounded by thinned cortex. |
|
|
|
Ewing Sarcoma
|
Malignant tumor occurring most frequently in long bones. It most frequently affects males between 10 and 20 years of age.
Intense local pain, swelling and pathological fractures. X-Ray : destruction of cortical bon with a periosteal reaction mainfesting with Codman triangle |
|
|
|
Radiologic manifestation of Ewing's sarcoma
|
Codman's triangle
|
|
|
|
Feared complication of acute mononucleosis and how to prevent it?
|
Splenic enlargment and subsequent splenic rupture.
Contact sports should be absolutely forbidden in this patient. In fact, strict bed rest, or even hospitalization, may need to be considered until the splenomegaly resolves. |
|
|
|
X-Linked genetic disease
|
"Disease" encoded by a gene on the X chromosome.
Males have a single X chromosome and are affected. Females have two X chromosomes and recessive X-linked disorders are rarely expressed in females. i.e. Duchenne's dystrophy |
|
|
|
Trisomy 21
- Disease - Incidence - Prognosis - Management |
Down syndrome
1:600 births Most common abnormality of autosomal chromosomes Rises with advanced maternal age from 1:2000 at age 20 to 1:20 by age 45. Prognosis : long term Management : - Recommend chromosomal analysis - Echo, thyroid test, atlanto-occipital x-ray at 2 and 12 years, hearing test, ophthalmology assessment - Early intervention programs help children reach full potential. |
|
|
|
Trisomy 21
Cranium Eyes Ears Facial features MSK Cardiac GI GU CNS Other |
Cranium
- Mild microcephaly, flat occiput, 3rd fontanelle, brachycephaly Eyes - Upslanting palpebral fissures, inner epicanthal folds, speckled iris, refractive errors, acquired cataracts, nystagmus and strabismus Ears - Low set, overfolded upper helix, frequent AOM, hearing loss Facial features - Protruding tongue, large cheeks, low flat nasal bridge, small nose MSK - Short stature, excess nuchal skin, joint hyperflexibility Cardiac - AVSD GI - Duodenal, esophageal, anal atresia, TE fistula, Hirschsprung, Chronic constipation GU - Cryptocorchidism CNS - Hypotonia at birth - Low IQ, developmental delay, hearing problems - Onset of Alzheimer's disease at 40 Other - Simian (transverse palmar) crease - Clinodactyly and absent middle phalanx of the 5 th figner |
|
|
|
Trisomy 18
Cranium Eyes Ears Facial features MSK Cardiac GI GU CNS Other |
Edwards syndrome
Cranium - Mild microcephaly, prominent occiput Eyes - Microophtalmia, hypotelorism, iris coloboma, retinal anomalies Ears - Low set, malformed Facial features - Cleft lip/palate, small mouth, micrognathia MSK - Short stature, clenched fist with overlapping digits, hypoplastic nails, clinodactyly Cardiac - 60% GI - Hernia GU - Polycystic kidneys, cryptocorchidism CNS - Hypertonia Other - Small for gestational age - Rocker bottom feet |
|
|
|
Trisomy 18
- Disease - Incidence - Prognosis - Management |
Edwards syndrome
1:6000 Prognosis : 44% die in first month, 10% survive past 1 year (profound intellectual disability in survivors) |
|
|
|
Trisomy 13
- Disease - Incidence - Prognosis - Management |
1: 10 000
Prognosis : - 33% die in 1st month - 50% by 2nd month - 90% by 1 year from FTT - Profound intellectual disability in survivors |
|
|
|
Turner syndrome
- Genotype - Incidence - Phenotype - IQ & behaviour - Gonad & reproductive function - Diagnosis - Prognosis - Management |
Genotype
- 45X (most common) - Mosaic: 46XX with p deletion, 45 XO, 47 XXX Incidence - 1:4000 live female births Phenotype - Short stature, short webbed neck, low posterior hair line, wide carrying angle - Broad chest, widely spaced nipples - Lymphedema of hands and/or feet, cystic hygroma in newborn with polyhydramnios, lung hypolasia - Coarctation of the aorta, bicuspid aortic valve - Renal and cardiac malformations + increased risk of HTN - Less sever spectrum with mosaicism IQ : mildly deficient to normal intelligence Gonad and reproductive function : - Streak ovaries with deficient follicles, infertility, primary amenorrhea, impaired development of secondary sexual characteristics. Prognosis - Normal life expectancy if no complications - Increased risk of X-linked disease Management - Echo, ECG to screen for cardiac malformation - GH therapy for short stature - Estrogen replacement at time of puberty for development of secondary sexual characteristics. |
|
|
|
Genotype of turner syndrome
|
Genotype
- 45X (most common) - Mosaic: 46XX with p deletion, 45 XO, 47 XXX |
|
|
|
Noonan Syndrome
- Genotype - Incidence - Phenotype - IQ & behaviour - Gonad & reproductive function - Diagnosis - Prognosis - Management |
Genotype
- 46 XX and 46 XY - Autosomal dominant with variable expression - Higher transmission of affected maternal gene Incidence - 1:2000 male and female live births Phenotype - Certain phenotypic features similar to female with Turner syndrome - Short stature, webbed neck, triangular facies, hypertelorism, low set ears, epicanthal folds, ptosis - Pectus excavatum - Right sided congenital heart disease, pulmonary stenosis IQ : moderate intellectual disability Gonad - Delayed puberty Management - Affected males may require testosterone replacement therapy at puberty - Echo, ECG |
|
|
|
Klinefelter syndrome
- Genotype - Incidence - Phenotype - IQ & behaviour - Gonad & reproductive function - Diagnosis - Prognosis - Management |
Genotype
- 47 XXY - 48 XXXY, 49 XXXXY Incidence : 1:1000 live male births Phenotype - Tall, slim, underweight - No features of prepuberty - Postpuberty : male may suffer from developmental delay, long limbs, gynecomastia, lack of facial hair IQ : Mild intellectual disability + behavioural or psychitaric disroders (anxiety, shyness, antisocial) Gonad - Infertility due to hypogonadism / hypospermia Management : - Testosterone in adolescents |
|
|
|
Fragile X syndrome
- Genotype - Incidence - Phenotype - IQ & behaviour - Gonad & reproductive function - Diagnosis - Prognosis - Management |
Genotype :
- X linked - Genetic anticipation Incidence - 1:3600 males (most common genetic cause of intellectual disability in boys) - 1:6000 females Phenotype - Overgrowth : prominent jaw, forehead and nasal bridge - Long thin face with large protuberant ears, macroorchidism - Hyperextensibility, high arched palate - Complications : seizures, scoliosis, mitral valve prolapse IQ : mild to moderate intellectual disability - ADHD or autism Diagnosis - Cytogenetic studies : region on Xq which fails to condense during mitosis, fragile X marker - Molecular testing |
|
|
|
DiGeorges Syndrome
- Genotype - Clinical features |
Genotype
- Micro-deletions of 22q11 Clinical features : CATCH 22 - C : cyanotic CHD - A : anomalies (craniofacial anomalies, typically micrognathia and low set ears) - T : thymic hypoplasia "immunodeficiency" recurrent infections - C : Cognitive impairment - H : Hypoparathyroidism and hypocalcemia - 22q11 micro-deletions |
|
|
|
Prader-Willi syndrome
|
Genotype
- Lack of paternally imprinted genes on chromosome 15q11 - Due to deletion of paternal chromosome 15q11 or two maternal chromosomes 15s 1: 15000 Clinical features - H3O : hypotonia and weakness, hypogonadism, obsessive hyperphagia, obesity - Short stature, almond shaped eyes, small hands and feet with tapering of fingers - Development delay - Hypopigmentation, DB2 |
|
|
|
Angelman Syndrome
|
Lack of maternally imprinted genes on chromosome 15q11
Clinical features: - severe intellectual disability, seizures, tremulousness - midface hypoplasia, fair hair, uncontrollable laughter |
|
|
|
Causes of increased CK
|
Prolonged exercice
Trauma Rheumatoid arthritis DMD (increased from birth) |
|
|
|
Cause of Duchenne's muscular dystrophy
|
1:4000 males
1/3 spontaneous mutations 2/3 X linked recessive Missing structural protein dystrophin --> muscle fibre fragility --> fibre breakdown --> necrosis and regeneration. |
|
|
|
Clinical features of Duchenne's muscular dystrophy
|
Proximal muscle weakness by age 3
Gower's sign : child uses hand to climb up, the legs to move from a sitting to a standing position Waddling gait Toe walking Hypertrophy of calf muscles and wasting of thigh muscles Decreased reflexes Dystrophin is expressed in the brain and boys with DMD may have delayed motor and cognitive development. Cardiomyopathy Moderate intellectual compromise |
|
|
|
Diagnosis of Duchenne's muscular dystrophy
|
Family history (pedigree)
Increased CK (50-100x normal) and LDH Muscle biopsy EMG |
|
|
|
Complications of Duchenne's muscular dystrophy
|
Patient usually wheelchair bound by 12 years of age
Early flexion contractures, scoliosis, develop osteopenia of immobility, increased risk of fracture Death due to pneumoniae / respiratory failure or CHF in 2nd or 3rd decade |
|
|
|
Treatment of Duchenne's muscular dystrophy
|
Supportive
- physiotherapy - wheelchairs - braces - prevent obesity Surgical (scoliosis) Use of steroids (prednisone or deflazacort) |
|
|
|
Picture of coloboma
|
|
|
|
|
Becker's muscular dystrophy
|
X-linked recessive due to a mutation in dystrophin gene with some protein production.
Symptoms similar to Duchenne but onset is later in childhood and progression is slower. Same management, investigations. Death later in 4th decade. |
|
|
|
VACTERL association
|
Should be suspected in all children with tracheo-esophageal fistula
- V : Vertebral dysgenesis 70% - A : Anal atresia (imperforate anus) +/- fistula 80% - C : Cardiac anomalies - VSD 55% - T-E : Tracheo-esophageal fistula +/- esophageal atresia 70% - R : Renal anomalies 55% - L : Limb anomalies - radial dysplasia, pre-axial polydactyly 55% May also present with a single umbilical artery or FTT |
|
|
|
CHARGE association
|
CHARGE
- C : Coloboma - H : congenital Heart disease - A : choanal Atresia - R : mental Retardation - G : GU anomalies - E : Ear anomalies Etiology thought to be due to abnormal development rather than a genetic mechanism. |
|
|
|
Treatment of VSD
|
Treatment of CHF
- Digitalis - Diuretics Surgical closure by 1 year of age |
|
|
|
Relationship between heart murmur and size of VSD
|
Size of VSD is inversely related to intensity of murmur.
|
|
|
|
Patent ductus arteriosus definition and normal ductus arteriosus physiology
|
Patent vessel between descending aorta and left pulmonary artery.
Ductus Arteriosus ________________________________ Functionnal closure within 15 hours of life, anatomical closure within first days of life. Delayed closure of ductus is normal in preterm infants Spontaneous closure is common in premature infants, less common in term infants. |
|
|
|
Presentation of patent ductus arteriosus
|
History : may be asymptomatic or have breath holding spells + poor feeding.
- If severe, can lead to CHF 10 % of congenital heart defects L > R shunt P/E : - heavy machinery murmur - high pulse rate - wide pulse pressure - hyperactive precordium - big pounding pulse |
|
|
|
Investigations of patent ductus arteriosus
|
ECG : may show LVH, LAH, BVH
CXR : increased pulmonary vasculature + prominent pulmonary artery Diagnosis by ultrasound |
|
|
|
Treatment of patent ductus arteriosus
|
Indomethacin - PGE1 antagonist in premature infants if necessary
Catheter or surgical closure if PDA is contributing to respiratory compromise, poor growth or persists beyond 6th month of life. |
|
|
|
What is an atrioventricular canal?
|
Type I ASD
High VSD Common atrioventricural valve |
|
|
|
Presentation of the coarctation of the aorta
|
Usually asymptomatic, if severe, presents with shock
- Differential cyanosis (Lower ext.), oliguria, neurologic symptoms, claudications....etc. 8% of congenital heart defects Commonly associated with : - bicuspid aortic valve (50%), Turner syndrome (35%) P/E : Upper extremities hypertension / Lower extremities hypotension Radial-Femoral delay Absent or systolic murmur with late peak at apex, left axilla and left back Prognosis : CHF Complicated hypertension |
|
|
|
Investigations of coarctation of the aorta
|
ECG : RVH early in infancy + LVH later in childhood
|
|
|
|
Treatment of coarctation of the aorta
|
Give prostaglandins to keep ductus arteriosus patent for stabilization :
1- balloon arterioplasty 2- surgical correction in symptomatic neonate If symptomatic or critically ill |
|
|
|
Treatment of aortic or pulmonic stenosis?
|
Surgical repair
|
|
|
|
Method to differentiate between cardiac and non cardiac cyanosis
|
Hyperoxic Test
1) Obtain preductal, right atrial ABG in room air 2) Repeat ABG after the child inspires 100% oxygen If PaO2 improves to greater than 150 mmHg, cyanosis is less likely cardiac in origin. |
|
|
|
Prevalence of aortic/pulmonic stenosis
|
5% each
|
|
|
|
Ebstein's anomaly picture
|
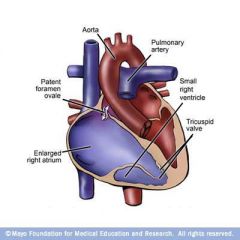
|
|
|
|
Transposition of great vessels picture
|
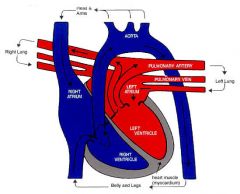
|
|
|
|
Treatment of the transposition of great arteries
|
1- Correct acidosis
2- Give prostaglandins PGE1 (Prostin) infusion to keep ductus open 3- Perform emergency septostomy or surgery (arterial switch procedure) Infants without VSD must be repaired within 2 weeks to avoid weak LV muscle |
|
|
|
Tetralogy of Fallot anomalies
|
Hypoplasia of the conus causing :
1. VSD 2. Right ventricular outflow obstruction 3. Aortic root "overriding" VSD 4. Right ventricular hypertrophy |
|
|
|
Prevalence of TOF
|
10%
Most frequent cyanotic cardiac anomaly |
|
|
|
Presentation of tetralogy of Fallot
|
Infants may initially have a L>R shunt and therefore are not cyanotic but the RVOTO is progressive, resulting in increasing R>L shunting with hypoxemia and cyanosis.
History : hypoxic "tet" spells - Primary pathology is hypoxia, leading to increased pulmonary vascular resistance and decreased systemic resistance, occuring in exertional states (e.g. crying, exercice) - Paroxysms of rapid and deep breathing, irritability and crying - Hyperpnea, increasing cyanosis often leading to deep sleep and decreased intensity of murmur (6 MO) - Peak incidence at 2-4 months - Children may have squatting If severe, may lead to seizures, loss of consciousness, death (rare) |
|
|
|
Management of tetralogy of Fallot hypoxic spells
|
1) Put child in knee-chest position (increases peripheral resistance which reduces R>L shunt and increases cerebral perfusion)
2) Administer O2 if uneffective : 3) Propranolol or morphine if uneffective : 4) Fluid bolus or phenylephrine Correct acidosis and anemia if present |
|
|
|
Investigations of TOF
|
ECG : RAD, RVH
CXR : boot shaped heart, decreased pulmonary vasculature, right aortic arch Heart U/S : it shows malformations |
|
|
|
Ebstein's anomaly
|
Congenital defect of the tricuspid valve in which the septal and posterior leaflets are malformed and displaced into the RV leading to variable degrees of RV dysfunction, TS, TR or functionnal pulmonary atresia if RV unable to open pulmonic valves.
RA massively enlarged, interatrial communication often exists allowing R>L shunting Often associated with WPW (accessory pathway) Associated with maternal lithium use and benzodiazepine during 1st trimester |
|
|
|
Treatment of Ebstein's anomaly
|
In newborns : consider closure of tricuspid valve + aortopulmonary shunt, or transplantation
In older children : tricuspid valve repair or valve replacement |
|
|
|
Transposition of great arteries
|
3%-5% of all congenital cardiac lesions (most common cyanotic CHD in neonate)
Parallel pulmonary and systemic circulation : - systemic : body - RA - RV - Aorta - systemic circulation - pulmonary : lungs - LA - LV - pulmonary artery - lungs |
|
|
|
Presentation of transposition of the great arteries
|
Newborn presents with progressive cyanosis unresponsive to oxygen therapy as the ductus arteriosus closes and mixing between the two circulation diminishes; severe hypoxemia, acidosis and death can occur rapidly.
if VSD present, cyanosis is not prominent and infant presents with CHF after a few weeks of life. The presence of an ASD or VSD or patent ductus arteriosus is essential for life. |
|
|
|
Investigations of TGA
|
ECG : RAD, RVH
CXR : egg-shaped heart with narrow mediastinum |
|
|
|
Truncus Arteriosus
|
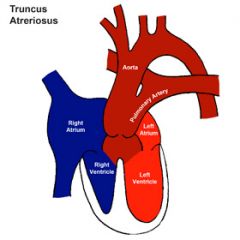
|
|
|
|
Hypoplastic left heart syndrome
|
1-3% of all congenital cardiac lesions.
Combination of : 1- Hypoplastic LV 2- Narrow mitral/aortic valves 3- Small ascending aorta 4- Contracted aorta with resultant systemic hypoperfusion Systemic circulation is dependant on ductus patency; upon closure of the ductus, infant presents with circulatory shock and metabolic acidosis. |
|
|
|
Treatment of hypolastic left syndrome
|
1- Intubate and correct metabolic acidosis
2- IV infusion of PGE1 to keep ductus open 3- Surgical correction (overall survival 50% to late childhood) or heart transplant |
|
|
|
Total anomalous pulmonary venous connection
|
Characterized by all of the pulmonary veins draining into the right sided circulation (supra-cardiac - SVC or innominate vein, infracardiac - Hepatic / portal vein or IVC, intracardiac, coronary sinus or RA
An ASD must be present to allow blood to shunt into the LA and systemic circulation. Treatment : surgical repair if severe cyanosis or CHF to pulmonary venous obstruction. |
|
|
|
Truncus Arteriosus
|
A single great vessel arising from the heart which gives rise to the aorta, PA and coronary arteries.
The truncal valve overlies a large VSD Treatment : surgical repair within first 6 months of life to prevent development of pulmonary vascular disease. |
|
|
|
Congestive Heart Failure etiologies in infants
|
Congenital Heart Disease
Arteriovenous malformations Cardiomyopathy Arrhythmias Acute hypertension Anemia Cor Pulmonale Myocarditis |
|
|
|
Clinical presentation of CHF in infants and key features.
|
4 Key features in congestive heart failure (2 tachy's 2 Megaly's)
1- Tachypnea 2- Tachycardia 3- Cardiomegaly 4- Hepatomegaly Presentation - Infant : feeding difficulties, easy fatiguability, exertional dyspnea, diaphoresis when sleeping or eating, respiratory distress, cyanosis, FTT - Child : decreased exercice tolerance, fatigue, decreased appetite, FTT, respiratory distress, frequent URTI's or asthma episodes Orthopnea, PND, pedal/dependant edema are all uncommon in children |
|
|
|
Management of congestive heart failure in infants
|
Correction of underlying disorders
General : sitting up, O2, sodium and water restriction, increased caloric intake Pharmacological : Digitalis, Diuretics, afterload reducers |
|
|
|
Which pediatric patients should receive antibiotic prophylaxis for infective endocarditis?
|
Cyanotic congenital heart disease
Rheumatic valve lesions (except if no valve involvement) Prosthetic heart valves Palliative shunts and conduits Previous endocarditis Pacemaker leads Amoxicillin 50mg/kg 30 to 60 minutes before procedure |
|
|
|
Most frequent sustained dysrythmia in children
|
Supraventricular tachycardia
|
|
|
|
Complete heart block
|
Congenital heart block can be caused by maternal Rho or La antibody
Often diagnosed in utero Symptomatic patients may require pacemaker |
|
|
|
Causes of vomiting in the newborn period
|
Incidence : 1:4500
Tracheoesophagal fistula Pyloric stenosis Duodenal atresia Malrotation of the intestine |
|
|
|
Presentation of tracheoesophagal fistula
|
May present after several months with vomiting, coughing and gagging.
Cyanaosis with feeds, respiratory distress, recurrent pneumonia Frothy bubbles of mucus in mouth and nose that return after suctionning Associated anomalies : VACTERL |
|
|
|
Management of tracheoesophageal fistula
|
1) Investigate for other congenital malformations
2) Early repair by surgical ligation to prevent lung damage and maintain nutrition and growth |
|
|
|
3 P's of pyloric stenosis
|
Palpable mass
Peristalsis visible Projectile vomiting (2-4 weeks after birth) |
|
|
|
Presentation of pyloric stenosis
|
Incidence : 1:500 M:F 4:1
Clinical Features : - non bilious projectile vomiting that ocurs after feeding - usually starts at 2-4 weeks of age - infant hungry and alert, will re-feed - dehydration, may lead to prolonged jaundice - gastric peristalsis goes from left upper quadrant to epigastrum - olive sign : olive shaped mass at margin of right rectus abdominus muscle - hypochloremic metabolic alkalosis |
|
|
|
Investigation of pyloric stenosis
|
- clinical
- U/S - X-Ray |
|
|
|
Presentation of duodenal atresia
|
Incidence 1:10 000 50% Premature
Clinical features : - bile stained vomiting if atresia distal to bile duct - no abdominal distention - dehydration - associated with Down syndrome, pematurity - often have history of maternal polyhydramnios |
|
|
|
X-Ray of duodenal atreisa
|
Double bubble sign
|
|
|
|
Treatment of duodenal atresia
|
Decompression with NG tube
Correction of metabolic abnormalities Surgical Correction |
|
|
|
Presentation of malrotation of the itnestine
|
Incidence 1:500, symptomatic 1:6000
80% experience symptoms in first two months of life. 3 presentations : 1- recurrent vomiting 2- FTT with vomiting 3- sudden onset abdominal pain and then shock Distented abdomen Vomiting due to volvulus and bands across duodenum |
|
|
|
Complication of malrotation of the intestine
|
Volvulus
Surfical emergency. It can result in bowel perforation, peritonitis and bowel necrosis. |
|
|
|
Causes of vomiting after the newborn period
|
Infectious and infalammatory
- GI causes `gastroenteritis, peritonitis, appendicitis, hepatitis, ulcers, pancreatitis, cholecystitis - Non GI causes : UTI, pyelonephritis, nephrolithiasis, otitis media, labyrinthitis, meningitis, pneumonia Anatomic - GI tract obstruction : intussusception, volvulus, foreign body GERD CNS - Increased ICP - Drugs/toxins - Migrain, cyclic vomiting Others - Metabolic/endocrine : DKA, inborn errors of metabolism, liver failure - poisons / drugs : lead, digoxin, erythromycine, theophylline - psychogenic - food alergy - overfeeding |
|
|
|
When are investigations required for infant GERD?
|
FTT
Recurrent cough Pneumonia GI blood loss Persistance of symptoms after 18 months |
|
|
|
Complications of GERD
|
Esophagitis
Strictures Barrett's esophagus FTT Aspiration |
|
|
|
Etiologies of intellectual disability
|
Genetic
- Down syndrome - Fragile X - PKU - Developmental brain anomalies - Inborn errors of metabolism Prenatal - Rubella - Fetal alcohol syndrome - Prenatal exposure to heroin - Cocaine - TORCH - HIV - Maternal DB - Maternal malnutrition - Birth trauma, hypoxia Perinatal - Prematurity - Low birth weight - Cerebral ischemia - Intracranial hemorrhage - Kernicterus Childhood - Intracranial infection - Head trauma - FTT - Lead poisonning Psychosocial factors : mild MR associated with low socioeconomic status, limited parental education, parental neglect, teen pregnancy, family instability |
|
|
|
Diagnosis of intellectual disability
|
Onset before 18 years of age of :
Below average general intellectual functioning as defined by an IQ of approximately 70 or below (2 standard deviations below the mean) AND deficits in adaptive functioning in at least two of : - Communication - Self-care - Home-living - Social skills - Self direction - Academic skills - Work - Leisure - Health - Safety Classification of intellectual disability (by IQ measurements) Mild 50-70 Moderate 35-49 Severe 20-34 Profound < 20 |
|
|
|
Management of intellectual disability
|
Main objective : enhance adaptive functioning level
Therapy : - emphasize community based treatment vs. institutionalization and early intervention - individual / family therapy, behaviour modification, multidisciplinary rehabilitation, medications for associated conditions Educations : - Life skills, vocational training, communication skills, family education Psychosocial support of parents and respite care |
|
|
|
Differential diagnosis of language delay
|
Hearing impairment
Cognitive disability - global developmental delay, mental retardation Pervasive developmental disorder, including autism Selective mutism Landau-Kleffner syndrome (acquired epileptic aphasia) Mechanical problems - cleft palate - cranial nerve palsy Social deprivation |
|
|
|
Risk factors for sensorineural hearing loss
|
Genetic syndrome - Family history
Congenital infections (TORCH) Craniofacial abnormalities < 1500 g birthweight Hyperbilirubinemia / kernicterus Asphyxia / Low Apgar scores Bacterial meningitis / viral encephalitis Language development may seem normal for up to 6 months but may regress de to lack of feedback. |
|
|
|
How to evaluate hearing loss in children
|
< 6 months : auditory brainstem response, tympanometry
> 6-8 months : behaviour audiometry > 3-4 old : pure tone audiometry |
|
|
|
What is selective mutism
|
A childhood anxiety disorder with onset age 5-6
Only speaks in certain situations, usually at home Healthy children with no hearing impairment Often above average intelligence |
|
|
|
Landau-Kleffner syndrome
|
Acquired epileptic aphasia
Present in late preschool to early school age years, may be similar to autism. Child begins developing language normally, then sudden regression of language |
|
|
|
Management of language delay
|
ENT and dental referral if mechanical cause
Speech therapy in disorders of fluency, receptive or expressive language Psychiatric consultation in selective mutism, PDD |
|
|
|
Definition of learning disorder and diagnosis
|
A specific and persistent failure to acquire academic skills despite conventional instruction, adequate intelligence and sociocultural opportunity
A significant discrepancy between a child's intellectual ability and his or her academic performance. ____________________________________________________________________________ Diagnosis : Individual scores on achievement tests in reading, mathematics or written expression (WISC III, WRAT) significantly below (> 2 SD) that expeted for age, education and IQ. Interferes with academic achievement or ADLs that require reading, mathematics or writing skills. |
|
|
|
Psychiatric comorbidities of a learning disorder
|
Dysthymia
Conduct disorder Major depressive disorder Oppositional defiant disorder ADHA |
|
|
|
What is fetal alcohol spectrum disorder?
|
The disorder refers to the full range of problems resulting from the use of alcohol during pregnancy, including Fetal Alcohol Syndrome and Fetal Alcohol Effects
|
|
|
|
Fetal alcohol syndrome diagnosis
|
Criteria for diagnosis of fetal alcohol syndrome :
1- Growth deficiency - low birth weight and / or length at birth that continues through childhood 2- Abnormal craniofacial features - small head, short palpebral fissures, long smooth philtrum, thin upper lip 3- CNS dysfunction - microcephaly and / or neurobehavioural dysfunction 4- Strong evidence of maternal drinking during pregnancy Cardiac and renal defects, hypospadia may occur. FASD is one of the most common causes of mental retardation in the world. |
|
|
|
Diagnostic criteria for daibetes mellitus in children
|
Same as adult
|
|
|
|
Management of uncomplicated type I diabetes in children.
|
Insulin injections 2-3 times per day, blood glucose monitoring.
Young children more susceptible to CNS damage with hypoglycemia with fewer benefits from tight control, hence target glucose range higher at 6-12 mmol/L Increasingly tighter control in older children 4-8 mmol/L |
|
|
|
Complications of type I diabetes in children
|
Hypoglycemia
- seizures - coma Hyperglycemia DKA Long term complications : - Retinopathy - Nephropathy - Neuropathy - Metabolic syndrome - Other auto-immune conditions : hypothyroidism, celiac disease |
|
|
|
Acute treatment of hypoglycemia in children
|
Glucagon injection
|
|
|
|
Blood glucose targets by Age
|
< 6 5.6-10 HbA1C 7.5-8.5%
6-12 5.0-10.0 HbA1C < 8% > 12 5.0-7.2 HbA1C < 7.5% |
|
|
|
Which oral hypoglycemian is preferred with children with type II diabetes? Why?
|
Metformin because it does not cause hypoglycemia
|
|
|
|
Causes and management of diabetes insipidus and SIADH in children?
|
Same as adults
|
|
|
|
Causes and management of diabetes insipidus and SIADH in children?
|
Same as adults
|
|
|
|
Causes of congenital hypothyroidism
|
Malformation of the thyroid gland (agenesis or ectopic)
Maternal factors : - iodine deficiency - prenatal exposure to antithyroid drugs or radioiodine |
|
|
|
Diagnosis of congenital hypothyroidism
|
Newborn screening with TSH and T$
|
|
|
|
Presentation of congenital hypothyroidism
|
Prolonged jaundice
Constipation Sluggish, hoarse cry, lethargy, poor feeding Macroglossia, coarse facial features, large fontanelles, umbilical hernia |
|
|
|
Management and prognosis of congenital hypothyroidism
|
Management : Thyroxine replacement
Prognosis : Excellent if treated within 1-2 months of birth If treatment started after 3-6 months of age may result in permanent developmental delay and / or mental retardation (mild to profound) |
|
|
|
Presentation of acquired hypothyroidism in children
|
Delayed bone age, FTT, short stature, goiter
Sexual pseudoprecocity : early sexual development with short stature and delayed bone age. Most commonly Hashimoto's thyroiditis Does not cause permanent developmental delay. Treated with 10 ug/kg/day |
|
|
|
Craniosynostosis
|
Craniosynostosis is a condition in which one or more of the fibrous sutures in an infant skull prematurely fuses by ossification,[1] thereby changing the growth pattern of the skull
|
|
|
|
Congenital hyperthyroidism presentation
|
Tachycardia with congestive heart failure, irritability, craniosynostosis, poor feeding, failure to thrive, heart murmur, goitre.
Usually caused by transplacental pasasge of maternal TSI's Spontaneous resolution by 2-3 months of life as antibodies cleared |
|
|
|
Management of congenital hyperthyroidism
|
PTU until antibodies cleared
|
|
|
|
Grave's disease in children
|
Same management as adults. May also present by school problems and personality changes. -
|
|
|
|
Management of thyroid nodule in children
|
Prompt evaluation as 30-40% have carcinoma
|
|
|
|
Presentation of congenital adrenal enzyme deficiencies
|

|
|
|
|
Etiology of ambiguous genitalia
|
Male Pseudohermaphrodite (XY)
- inborn error of testosterone biosynthesis or Leydig cell hypoplasia - 5-alpha-reductase deficiency, androgen receptor deficiency or insensitivity - LH / hCG unresponsiveness - small phallus, hypospadias, undescented testicles Female Pseudohermaphrodite (XX) - virilizing congenital adrenal hyperplasia - maternal source : virilizing ovarian or adrenal tumours, untreated CAH, placental aromatase deficiency -virilization of external genitalia : clitoral hypertrophy, labioscrotal fusion Mixed pattern True hermaphrodite - both ovarian follicles and seminiferous tubules in the same patient with a 46XX karyotype - increased risk of malignant transformation of gonad tissue Mixed gonadal dysgenesis |
|
|
|
Investigations for ambiguous genitalia
|
Karyotype
Electrolytes and renin (CAH) Measurement of 17-OH-progesterone, androgens, FSH and LH Abdominal U/S to look for uterus, testicles, ovaries |
|
|
|
Most common cause of ambiguous genitalia
|
Congenital Adrenal Hyperplasia
|
|
|
|
Transmission of congenital adrenal hyperplasia
|
Autosomal recessive condition causing partial or total enzyme defect.
|
|
|
|
Most common deficiency with congenital adrenal hyperplasia
|
21 alpha hydroxylase
|
|
|
|
Pathophysiology behind congenital adrenal hyperplasia.
|
Enzyme deficiency resulting in decreased secretion of cortisol which raises ACTH levels causing the hyperplasia.
Symptoms depend on the enzyme deficiency. |
|
|
|
21 Hydroxylase deficiency types
|
1) Late onset (mild)
- girls present with amenorrhea - boys present with precocious puberty with early adrenarche, dehydration - Accelerated linear growth in early puberty but early fusion of epiphysis leading to decreased adult height. - Dx : increased plasma 17-OH-progesterone after ACTH stimulation test 2) Salt wasting (2/3 of cases) - infants present with shock, FTT, low Na, high K, low Cl, low glucose, high ACTH - hyperpigmentation of genitals and areola 3) Simple virilizing 21 hydroxylase deficiency - virilization in girls |
|
|
|
Management of salt wasting 21 hydroxylase deficiency
|
Replace fluids and electorlytes
Manage hypoglycemia Hydrocortisone Mineralocoritcoids Surgery to correct ambiguous genitalia |
|
|
|
Investigations for congenital adrenal hyperlasia
|
CBC
Electrolytes Renin 17-OH-progesterone (substrate for 21 hydroxylase) ACTH stimulation test Cortisol Testosterone, DHEAS, urinary 17-ketosteroids bone age |
low Na, high K, low cortisol, high ACTH,
increased 17-OH-progesterone Increased testosterone, DHEAS, urinary 17-ketosteroids Advanced bone age |
|
|
Etiology of central precocious puberty
|
Hypergonadotropic hypergonadism (Premature activation of the Hypothalamo-gonadal axis)
- Idiopathic or constitutionnal - CNS disturbances : tumour, hamartoma, post-meningitis, increased ICP, radiotherapy - Neurofibromatosis - Primary severe hypothyroidism A child with proven central precocious puberty should receive an MRI of the brain. |
|
|
|
Etiology of Pseudo (peripheral) precocious puberty
|
Hypogonadotropic Hypergonadism
- Adrenal disorders : CAH, adrenal neoplasm - Testicular / Ovarian tumour - Gonadotropin secreting tumour: hepatoblastoma, intracranial teratoma, germinoma - Exogenous steroid administration - McCune-Albright syndrome |
|
|
|
McCune Albright Syndrome
|
Precocious puberty
Café au lait spots Fibrous dysplasia of skeletal system |
|
|
|
Causes of delayed puberty
|
Hypogonadotropic Hypogonadism
- Constitutionnal - Genetic syndromes (e.g. Kallman) - Tumours Hypergonadotropic Hypogonadism - Genetic (Turner, Kleinefelter) - Gonadal damage - infection, radiation, trauma - Gonadal dysgenesis - Hormonal defect - Androgen insensitivity, 5 alpha reductase deficiency |
|
|
|
Investigations for delayed puberty
|
Hormone levels : estradiol, testosterone, LH, FSH, TSH, GnRH, test bone age
Consider MRI of the head Karyotype |
|
|
|
Definition of precocious puberty
|
Secondary sexual development
< 8 years in girls < 9 years in boys More common in females, more worrisome in males (higher incidence of pathology) |
|
|
|
Indications for medical intervention to delay progression of puberty
|
Male sex
Age < 6 Bone age advancing more quickly than height age Psychosocial issues |
|
|
|
Rx to delay central precocious puberty
|
GnRH agonists
e.g. Leuprolide |
|