![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
71 Cards in this Set
- Front
- Back
|
Pyridoxine dependent epilepsy 1. what can be found in high levels in CSF? 2. When sz. can start? 3. Type of sz. 4. clinical characteristics other than sz. |
1. Pyridoxal 5 phosphate 2. hours -days from birth, but range from in-utero to months from birth 3. erratic myoclonic convulsions, but can be convulsive SE. 4. irritability, sleeplessness, abnormal eye movements, facial grimacing. Might suggest HIE. 5. May include non specific brain malformations: partial ACC, mega cisterna magna.. |
|
|
Tx. in pyridoxine dependent epi. |
single dose of pyridoxine: 50-100mg, responders are at risk for prolonged apnea or stupor, thus- f/u for 48 h is mandatory. \ Sz. stops and EEG (usually burst-suppression) is normalized in 20 min. maintenance tx. 15-30 mg/kg /day div. 2-3 doses, not exceed 500 mg to prevent pyridoxal toxicity. Consider adding folinic acid if response is partial |
|
|
List 8 parameters suggesting inherited metabolic epilepsy |
1. consanguinity 2. onset in the neonatal period 3. poor feeding 4. lactic acidosis 5. myoclonic sz. 6. apneic spells 7. EEG: burst suppression, generalized spike-wave 8. poor response to traditional AED |
|
|
Biomarkers for Pyridoxine dependent epi |
elevated levels of alpha-AASA & pipecolic acid eleveated levels of pyridoxal 5 phosp. in CSF mutation analysis of ALDHTA1 |
|
|
Other etiologies responsive to pyridoxine? |
1. infantile spasm 2. KCNQ2 mutations epi. |
|
|
The role of biotin and biotinidase |
Biotin is a cofactor for 5 carboxylation enzymes, e.c. pyruvate carboxylase, acetyl coA carboxylase, etc. Biotinidase releases the biotin from the protein it bounded with. |
|
|
Biotinidase def. - characteristics: |
1. T-C or myoclonic or IS sz. 2. irritability, conjunctivits, cheilosis, alopecia. 3. By 1 y ageL optic atrophy and SNHL 4. EEG: range from normal to burst suppression 5. MRI - diffuse WM changes, striatal edema |
|
|
Tx. of biotinidase def. |
5-20mg of biotin once daily |
|
|
Glut-1 def. characteristics |
1. Glu in CSF <40 or Glu ration <0.4 CSF:serum 2. 90% of pt. develop epi, most common early onset abscence 3. EEG shows focal or generalized attenuation but may be normal 4. MRI- cerebelar atrophy 5. microcephaly, ataxia, dev. delay |
|
|
Glut-1 def. therapy |
1. rapid response to ketogenic diet 2. trials to use anaplerotic agent: Trihepanotin 3. Avoid drugs that inhibit the transporter: Phenobarb, diazepam, theophylline and caffeine, alcohol, valporate. |
|
|
Cerebral folate def. - what molecule is low in the CSF? |
5MTHF May be primary due to mutation in folate transporter FR1 or secondary to Ab. Most common - end stage of various neurodegenerative disorders: Rett syn., mitochondrial dis... |
|
|
Classic cerebral folate def.? |
Intractable TC sz. in infancy or early childhood. Tx - folinic acid, cause folate does not cross BBB |
|
|
Thiamine respnsive basal ganglia dis. (included in Leigh syndrome) describe |
acute-sub acute encephalopathy accompanied by sz. described in Saudi-arabian pedgrees may start from neonatal period to adulthood. MRI- striatal, pallidal, thalamic T2 hyper intensities. Deteriorate to hypertensive encephalopaty and coma. Mutations in SLC19A3 thiamine transporter |
|
|
Indications for trial of Thiamine+ biotin: |
IS with basal ganglia involvment Leigh syn. Sz. with generalized dystonia Wernicke encephalopathy give 300-900mg thiamine with 5-10 mg/kg biotin |
|
|
Congenital microcephaly, poor development, refractory sz, and low or absent serine levels- diagnosis? tx. ? |
Impaired synthesis of L-serine due to mutated 3- phosphoglycerate dehydrogenase. Start therapy with serine, and improve outcome. May be given pre-natal in known cases |
|
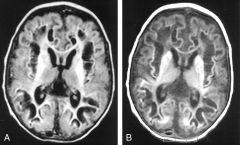
Myoclonic sz. started on day 3, accompanied by irritability and poor feeding. EEG: burst suppression. diagnosis? |
1. Sulfite oxidase or Molybdenum co-factor def. 2. lab: low uric acid and low plasma homocysteine. 3. MRI first days of life diffuse cerebral edema with psudo-cysts |
|
|
Novel therapy for Sulfite oxidase? |
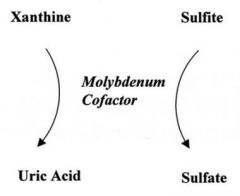
cyclic pyranopterin monophosphate - percusor of molybdenum |
|
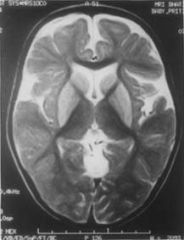
12 m male, hx. of profound MR + dystonia+ choreoathetosis, started @ 2 m of age after post vaccination febrile dis. Diagnosis? |
Glutaric aciduria type 1 1:100,000 Increased urine glutaric acid metabolites. Macrocephaly+ increased sub-acachnoid spaces and progressive extra-pyramidal features and encephalpathy. |
|
|
What AED should be avoided when suspecting Glutaric aciduria and why? |
Depalept. may exacerbate metabolic imbalance by affecting acetyl CoA/CoA ratio. |
|
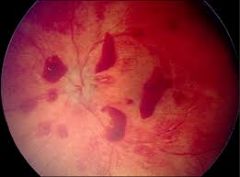
findings in fundoscopic examination of 7m baby girl, brought to ER due to lethargy. Describe. What else to look for? |
Retinal bleeding in various ages. The triad: retinal bleeding, subdural hematoma and acute encephalopathy delinate "shaken baby sydrome". Look for sub-dural hematoma and brain edema bone fractures skin laceration and bruises |
|
|
Describe the purposed mechanism for shaken baby syndrome: |
rapid and repetitive flexion, extension, and rotation of the head and neck around a relatively stable torso with or without impact injury. Shaking the baby's head forcefully while holding its torso in a pace of 2-5 times per second can cause Sub dural hematoma, intra-cranial tear of blood vessels and retinal bleeding as described above. |
|
|
Neurologic deterioration in abusive BI is the result of: (1 wrong) |
1. sub dural hematoma 2. Brain edema due to recurrent concussions of the gelationus brain against the firm vault 3. diffuse axonal injury 4. multiple tears in small intracranial blood vessels 5. disturbances in blood pressure autoregulation in the brain due to recurrent trauma 6. cervico-medullary injury with subsequent edema/ compression of respiratory center |
|
|
What is the specifity of retinal hemorrhages beyond the peri-natal period in detecting abusive BI? |
94%, and in cases where no signs of car trauma or multiple skull fractures: 100% |
|
|
3 dO baby girl presented with clonic rt. hand and rt. mid face rhythmic movements for 1-2 min. Hx. - non contributing, PE: bullous-erythematous rash over left limbs and lt. groin. Lab. tests: eosinophyllia - 6000cells suggested diagnosis? suggest a confirmatory test |
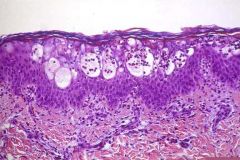
Incontinentia pigmenti (Bloch-Sulzberger dis.) Skin biopsy in the bullous stage=eosinophylic spongitis dermatitis. |
|
|
Incontinentia pigmenti- genetic? |
X-linked : Xp28 NEMO males with mutation do not survive, unless XXY karyotype or musaic. |
|
|
CNS involvement in the disease: |
in 30-50% of pt. 1. CNS microangiopathy and strokes 2. acquired microcephaly 3. mental retardation - 25% of pt. 4. cerebral dysplasia |
|
|
Clinical characteristics: |
Skin- 4 stages Teeth : 60% nails: 50% eyes: 35% CNS: 35% skeletal: 15% * eye manifestations goes together with CNS abnormalities * increased risk for AML, Wilms, retinoblastoma
|
|
|
Place the following structures on the picture: 1. frontal, parietal, temporal, occipital lobes 2. rolandic (central) and sylvian fissures 3. angular gyri, superior temporal sulcus |

|
|
|
describe the territory of the MCA, ACA and PCA |
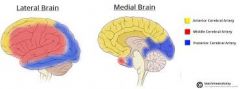
The external part of the cortex=MCA, the "inside" part = ACA |
|
|
what deficits apparent after occlusion of the Lt. vertebral artery at the level of post. cerebellar artery? What deficits appear after occlusion of the basilar artery at the level of the superior cerebellar artery? |
1. almost no deficits because both vertebral arteries supply the same structures 2. total blindness, cause both sides leave from the same artery and supply the visual cortex. |
|
|
Describe 4 main pathological pathways causing progressive neuro-degenerative disease |
1. accumulation of toxic substrate proximal to an enzymatic defect: Amino-acidemias, Organic acidemias, urea cycle, glucose and lipid metabolism. Identified through lab w/u 2. Deficiency in energy: mithocohondrial diseases, fatty acid oxidation, pyruvate cycle, transporters. requires stressogenic situation or biopsies to identify 3. Storage diseases and inability to catabolize/clean products from the cells 4. genetic defects, mechanism not clear |
|
|
Heteroplasmy- definition: |
Each cell has different number of mithocondria, with slightly different set of genes in each cell. |
|
|
Describe the utilization of glucose for energy in the cell: |
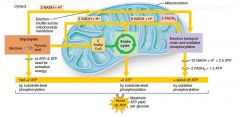
when no oxygen exist, then only 2 ATP + lactate is created by metabolism of pyruvate to lactate |
|
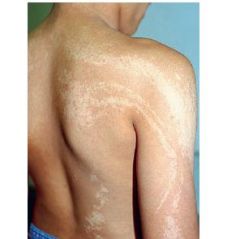
diagnosis? genetics? |
Hypomelanosis of Ito- 3rd most common phakomatose dis. Genetics: somatic musacism |
|
|
What is the most common neurodegerative genetic disorder? |
Neuronal lipo fucinosis |
|
|
Describe the path of auditory information |
The affarent system in the cochlea contains 3500 hair cells. All are connected with high speed axons to the cochlear nuclei in the medulla. There, and in 4 more relay stations it goes through a process of amplification, till it end in the 41st and 42nd auditory areas in the superior temporal gyrus. The rely stations are the superior olive, lateral leminscus, inferior colliculi, and medial geniculate body in the thalamus. |
|
|
What is the basic metabolic problem in NCL? |
Dysregulation of sphingolipids. In NCL1 the missing enzyme is a lysosomal enzyme Tpp1 which causes intracytoplasmic accumulation of saposin a and d. |
|
|
In which disease babies smell like cabbage? |
Tyrosinemia 1 |
|
|
What is the lab system employed to detect new born errors in metabolism and what incidence it idenity errors? |
Tandem mass spectrometry 1:2000-1:4000 |
|
|
What is the roll of supplemented glycine in acute presentation of IEM? |
It creares acylglycine that is non toxic and excreted in the urine easily |
|
|
Whats the roll of carnitine supplementation? |
It forms acylcarnitnes which reduces level of ketones and level of toxic organic acids creared in amino aciduria |
|
|
What is the connection between H1N1 vaccine and neuropathy? |
Suspected to cause GBS following massive immunization on 1976 and on 2009, but did not showed statistical correlation |
|
|
What substrate can be diagnosed in the urine following lead intoxication? |
Leuvinic acid can be found in the urine when lead levels in the blood are 40-40mg/dL |
|
|
What is the function of phenylalanine hydroxylase? |
Hepatic enzyme. Covert Phe to Tyrosine |
|
|
What are the normal levels of Phe,the hyperphenylalaninemia and PKU levels? |
Normal: 30-110micromol HyperPhenylalaninemia: 360-600 Phenylketonuria: more then 1000 |
|
|
Therapy for PKU |
Restriction of Phe from the diet and providing tyrosine suppl. |
|
|
What are the clinical signs of Lead toxiciity? |
1.motor mainly axonal neuropathy in LL More prevalent in adults 2. Encephalopathy more prevalent in kids 3. Colic abdominal pain 4. Pallor 5. Anemia microcytic hyopochromic |
|
|
Levels of Phe are related to: |
pAH activity in the liver |
|
|
Clinical manifestation of PKU |
1. Profound MR 2 acquired microcephaly 3. Hyeractive DTR 4. Hypotonia/spacticity 5. Light complex-hair,skin 6. Musty odor 7. Endpoint of athetotic CP 8. Adhd autistic features |
|
|
Congenital amyelinating neuropathy? |
Looks like congenital sma Born with artherogryphosis Dyspahgia and dyspnea Die early Extreme spectrum of Dejjerine sottas HMSN III |
|
|
what are the manifestations of hyperphenylalaninemic kids? |
Normal development |
|
|
A woman with classic PKU not treated, describe her baby: |
1. MR- 90℅ 2. Microcephaly-70℅ 3. Iugr-40℅ 4. Congenital heart defect - 12℅ |
|
|
Parinaud syndrome include? |
1. Up-gaze ophtalmoplegia 2. Poor convergence reaction with normal reaction to light 3. Vertical nystagmus |
|
|
Fazio-londe disease?? |
Progressive motor neuron damage of CN 9,10,12,5. AR, starts 1-10y,mutation in SLC52a Die in 9 m Pathology same as in sma |
|
|
Pathophysiology of PKU |
1. Decreases biosynthesis of myelin proteins secondary to excessive phe 2. Imbalance in the concentration of amino acids in the brain 3. Decreases levels of tyrosine and triphtophan- reduced levels of neurotransmitters |
|
|
Bilateral facial nerve pralysis suggests: |
GBS Mobius syndrome Traumatic labour with or w/o forceps Mobius 2nd to ergotamine teratogenic effect |
|
|
2 yO boy with dev.delay, hypotonia, mild ataxia, distal weakness, short stature and elevated protein in the CSF. Diagnosis? Pathology? |
Dejerines-sottas dis.- hypomyelination disease, more pns>cns. Peripheral nerves are very thick. Different mutations in myelin proteins |
|
|
What is the prognosis of kids with classic PKU treated with restriced phe diet? |
Normal IQ, problems with spelling, math, reading. Prone to depression, anxiety, phobia |
|
|
Can you breastfed a baby with classic phenylalanine? |
Yes |
|
|
How much phe should be provided in the diet of a child with classic pku? |
According to blood levels: 120-360 <12 y old 120-600 >12y old |
|
|
For a pt. With hyper phenylalaninemia-what diet recommendations? |
Keep levels of phe in blood same as classic pku only by protein restriction Dietry therapy for pregnant women |
|
|
Why mitochondrial disease cause extrnal ophtalmoplegia? |
The muscles of eye movements are full with mitochondria, more then any other muscle |
|
|
Chronic progressive external ophtalmoplegia can be part of: |
Mitochondrial disease Graves dis Oculopharyngeal muscular dystrophy Myesthenia gravis |
|
|
Christianson syn. |
Synonim: Angelman like syndrome MR, hypotonia, no speech, seizures Excessive drooling episodic laughter Gene-SLC19A6 syndromic ID x-linked Cerebellar atrophy in MRI |
|
|
Newborn 7 days old found positive on newborn screening test for c3: proprionic acidemia or secondary elevated pro.acid. Hx. Not contributing, fed on milk based formula. What might be the clinical signs? |
Vomiting, lethargy, hypotonia |
|
|
Abnormal lab results include: 1. Metabolic acidosis 2. Ketosis 3. Hyper amonemia 4. Neutropenia 5. Thrombocytopenia |
A. 1,3 B. 2,4,5 C. 1,4, 5 D. All |
|
|
What is the lab marker for homocystenuria? |
Methionine |
|
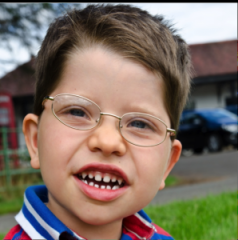
Severe encephalopathy |
Pit hopkins syndrome Post natal microcephaly Strabismus Epilepsy Stereotypic hand movements w/o loosing purposful hand movements Small hands and feets |
|
|
Proprionic acidemia-which organ may be damaged? Methyl malonic? |
Proprionic - cardiomyopathy Methyl malonic - renal |
|
|
Which type of Gaucher dis is prevalent in Ashkenazi jews? |
Type1- non neuropathic |
|
|
Spacity +bulbar dysphagia+ trismus+ head retraction in a baby |
Gaucher type 2=neuropathic crises |