Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
100 Cards in this Set
- Front
- Back
|
study of all genes and their interactions?
|
genomics
|
|
|
study of all proteins expressed in a cell?
|
proteomics
|
|
|
two types of strategies for isolating genes involved in clinical disorders?
|
1. functional
2. positional |
|
|
4 catagories of genetic disorders?
|
1. mendelian
2. cytogenetic 3. single-gene non-mendelian 4. multifactorial |
|
|
what are cytogenetic disorders?
|
numberic or structural changes in chromosomes
|
|
|
what type of cells are responsible for hereditary disease?
|
germ cells
|
|
|
loss (monosomy) or gain (trisomy) of whole chromosomes is known as?
|
genome mutations
|
|
|
abnormal numbers of chromosomes or alterations of chromosomal structure are known as?
|
chromosome mutations
|
|
|
a substitiution of one nucleotide for another?
|
point mutation
|
|
|
insertion or deletion of 1-2 base pairs?
|
frameshift mutation
|
|
|
what is the 6th amino acid (glu) of beta-globin replaced by in sickle cell disease?
|
valine
|
|
|
what type of mutation is the one AA switch that occurs in sickle cell disease?
|
point-mutaiton
|
|
|
the term applied to the situation where both alleles for a gene are fully expressed?
|
co-dominance
|
|
|
what does the term penetrance refer to when describing the expression of a trait caused by a mutant gene?
|
the percent expressing the trait of the total with the mutant gene
|
|
|
abnormal trait is present in all with mutant gene, but affected individuals show different clinical features?
|
variable expressivity
|
|
|
does loss of function for non-enzymatic vs enzymatic proteints effect heterozygotes equally?
|
no, LOF for enzymatic proteins is usually compensated for by normal allele, but LOF for non-enzymatic proteins are not thus disease ensues
|
|
|
of the mendalian disorders, the largest number are passed via what inheritance type?
|
autosomal recessive
|
|
|
what stage in life will most with AR pattern diseases be recognized?
|
early in life
|
|
|
what are the four mechanisms by which mendalian disorders occur?
|
1. altered structure of function of non-enzymatic proteins
2. defects in membrane receptors and transport systems 3. enzyme defects 4. mutations resulting in unusual reaction to drugs |
|
|
mutation of FBN1 on 15q21 leading to defective fibrillin-1 which further causes abnormal microfilbrils of elastic fibers?
|
Marfan syndrome
|
|
|
disorders in which collagen fibers are defective?
|
Ehlers-Danlos syndromes
|
|
|
all types of Ehlers-Danlos syndrome are AD except for which two AR types?
|
1. type VI
2. type VIIc |
|
|
defect in NF-1 gene on 17q11 causes what mendelian disorder?
|
neurofibromatoses types 1 and 2
|
|
|
Type 1 neurofibromatoses is also called?
|
von Recklinghausen disease
|
|
|
what are the 3 main clinical manifestations of NF type 1?
|
1. neurofibromas
2. cafe au lait spots 3. pigmented iris hamartomas (Lich nodules) |
|
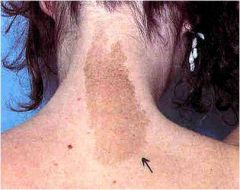
what do I have?
|
Neurofibromatoses type 1
|
|
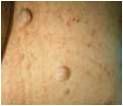
what do I have?
|
NF type 1
|
|
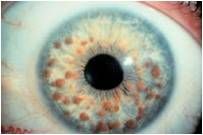
what do I have?
|
NF type 1
|
|
|
what disorder is characterized by a mutation in the genes encoding the merlin protein which regulates Schwann cell proliferation?
|
NF type-2
|
|
|
CNS tumors such as acoustic schwannomas can be seen with this genetic disorder?
|
NF type-2
|
|
|
mutation in the gene coding the receptor for LDL causes?
|
familial hypercholesterolemia
|
|
|
skin and tendinous xanthomas are seen with this Mendalian disorder?
|
familial hypercholesterolemia
|
|
|
what are 3 disorders which can be classified as lysosomal storage diseases?
|
1. tay-sachs disease
2. mucopolysaccharidosis I 3. Gaucher's disease |
|
|
caused by a deficiency in hexosaminidase A?
|
Tay Sachs Disease
|
|
|
characterized by a cherry red spot in macula by funduscopic exam?
|
Tay Sachs Disease
|
|
|
characterized by an accumulation of GM2 gangliosides?
|
Tay Sachs Disease
|
|
|
shows up arounf 6 months of age and kills child at 2 or 3 years of age?
|
Tay Sachs Disease
|
|
|
Progressive motor and mental deterioration including blindness, dimentia, and muscular flaccidity?
|
Tay Sachs Disease
|
|
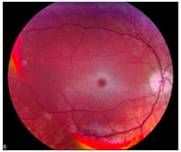
what do I have?
|
Tay Sachs Disease
|
|
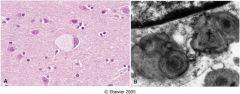
what do I have?
|
Tay Sachs Disease
|
|
|
what is the most common lysosomal storage disorder?
|
Gaucher's Disease
|
|
|
characterized by a defect in glucocerebrosidase?
|
Gaucher's Disease
|
|
|
characterized by an accumulation of glucocerebrosidase in phagocytes?
|
Gaucher's Disease type 1
|
|
|
characterized by an accumulation of glucocerebrosidase in cells of the CNS?
|
Gaucher's Disease type 2
|
|
|
which type of Gaucher's Disease appears in adulthood?
|
type 1
|
|
|
which type of Gaucher's Disease appears in childhood?
|
type 2
|
|
|
diagnosed via measured glucocerebrosidase activity in peripheral blood leukocytes or skin fibroblasts?
|
Gaucher's Disease
|
|
|
characterized by a deficiency in iduronidase?
|
mucopolysaccharidosis
|
|
|
the major type of mucopolysaccharidosis is called what?
|
Hurlers syndrome
|
|
|
hepatosplenomegaly by age 2, growth and mental retardation, coarse facial features, and death by age 10 from cardiovascular complications is characteristic of?
|
mucopolysaccharidosis
|
|
|
what are the 2 major types of glycogen storage disease?
|
1. hepatic
2. myopathic |
|
|
characterized by deficiency of muscle phosphorylase?
|
glycogen storage disease- McArdle syndrome
|
|
|
characterized by deficiency of glucose-6 phosphatase?
|
glycogen storage disease - hepatic types
|
|
|
muscle cramps after exercise, weakness, and myoglobin may be excreted as red urine in this disorder?
|
mucopolysaccharidosis - myopathic
|
|
|
generalized glycogenosis is called?
|
Pompe Disease
|
|
|
deficiency of acid maltase is characteristic of this disorder?
|
Pompe Disease - generalized glycogenosis
|
|
|
this disorder results in the storage of glycogen in lysosomes of all organs?
|
Pompe Disease - generalized glycogenosis
|
|
|
characterized by death at age 2 from massive cardiomegaly?
|
Pompe Disease - generalized glycogenosis
|
|
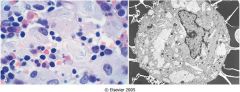
what do I have?
|
gauchers disease
|
|

what do I have?
|
Pompe Disease - generalized glycogenosis
|
|
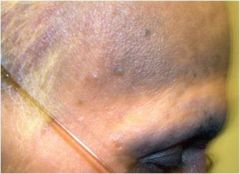
what do I have?
|
alkaptonuria
|
|
|
characterized by a deficiency in homogentisic oxidase?
|
alkaptonuria
|
|
|
characterized by crippling arthritis by ages 30-40?
|
alkaptonuria
|
|
|
the number of chromosomes is not a multiple of 23?
|
aneuploidy
|
|
|
loss of one chromosome due to nondisjunction during gametogenesis?
|
monosomy
|
|
|
gain of one chromosome due to nondisjunction during gametogenesis?
|
trisomy
|
|
|
two or more populations of cells in same individual?
|
mosaicism
|
|

what is this?
|
translocation
|
|
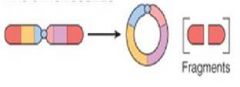
what is this?
|
Ring Chromosome
|
|
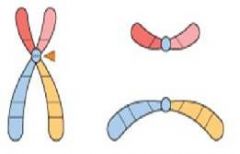
what is this?
|
Isochromosome
|
|
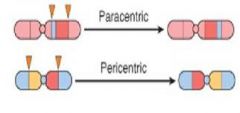
what is this?
|
Inversion
|
|
|
what is the most common chromosomal disorder?
|
trisomy 21
|
|
|
how does trisomy 21 occur?
|
nondisjunction during meiosis resulting in a gamete with an extra chromosome 21
|
|
|
characterized by congenital heart disease (40%), increased risk of acute leukemia (10-20 fold), and early onset Alzheimers disease (by age 40)?
|
Trisomy 21
|
|
|
what disorder is characterized by the deletion of band 11.2 on the long arm of chromosome 22?
|
DiGeorge syndrome or Chromosome 22q11.2 Deletion Syndrome
|
|
|
thymic hypoplasia and parathyroid hypoplasia make people with this disorder prone to infections and hypocalcemia?
|
DiGeorge syndrome or Chromosome 22q11.2 Deletion Syndrome
|
|
|
diagnosed by FISH probes and has a characteristic, large 1.5KB deletion of many unknown genes?
|
DiGeorge syndrome or Chromosome 22q11.2 Deletion Syndrome
|
|
|
the name of the inactivated X chromosome in women that is seem as a small dark body at the edge of the nucleus?
|
Barr body
|
|
|
disorder characterized by the presence of two or more X and one or more Y leading to reduced spermatogenesis and infertility?
|
Klinefelter Syndrome
|
|
|
47 XXY?
|
Klinefelter Syndrome
|
|
|
+ Increased length between soles and pubic soles(appearance of elongated body)
+ Small, atrophic testes; small penis, + Lack of secondary male characteristics (deep voice, beard, distribution of pubic hair) + Gynecomastia + Lower IQ, but mental retardation uncommon |
Klinefelter Syndrome
|
|
|
46,XY / 47,XXY
|
Klinefelter Syndrome
|
|
|
45,X
|
Turner Syndrome
|
|
|
+ Female hypogonadism; phenotypic females
+ May not be detected until puberty delayed: absent menses; no secondary sexual changes |
Turner Syndrome
|
|
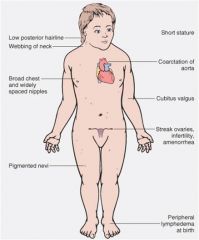
what do I have?
|
Turner Syndrome
|
|
|
What am I if I have the presence of both testicular and ovarian tissue?
|
True hermaphrodite
|
|
|
+ Genetic sex: always XX
+ Ovaries and internal genitalia normal + External genitalia: virilized (enlarged clitoris) + Cause: excessive exposure to androgenic steroids in early gestationk, often due to congenital adrenal hyperplasia (autosomal recessive trait) |
Female Pseudohermaphrodite
|
|
|
+ Possess a Y chromosome
+ Gonads are ftestes + Ducts and external genitalia: ambiguous or female (heterogeneous features) + Cause: defective virilization of male embryo |
Male Pseudohermaphrodite
|
|
|
most commone cause of Male Pseudohermaphrodites?
|
complete androgen insensitivity at androgen receptor
|
|
|
Second most commone cause of mental retardation after trisomy 21?
|
Fragile-X Syndrome
|
|
|
Clinical features worsen with progressive generations?
|
fragile-X syndrome
|
|
|
caused by long repeating sequences of three nucleotides due to mutation in familial mental retardation gene (FMR-1)?
|
fragile-X syndrome
|
|
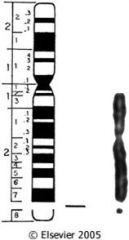
what do I have?
|
Fragile-X Syndrome
|
|
|
what encodes enzymes for oxidative phosphorylation?
|
maternal mitochodrial DNA
|
|
|
characterized by progressive bilateral loss of central vision which is first noted between 15 and 35 and leads to blindness. Cardiac conduction defects may also be present.
Pedigree: |
Leber Hereditary Optic Neuropathy
|
|
|
characterized by mental retardation, short stature, obesity, small hands and feet, and hypogonadism?
|
Prader Willi Syndrome
|
|
|
caused by deletion of q12 band on the paternal chromosome 15?
|
Prader Willi Syndrome
|
|
|
characterized by mental retardation, ataxic gait, seizures, and inappropriate laughter (“happy puppets”)?
|
Angelman Syndrome
|
|
|
what are two cytogenentic anlysis methods availible for studying chromosomes?
|
1. karyotyping
2. Flourescence in-situ hybridization (FISH) |
|
|
what are two molecular analysis methods for studying DNA?
|
1. PCR
2. Linkage Analysis |