Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
185 Cards in this Set
- Front
- Back
|
1. Four different stages of development in which children are prey to disorders *that we have data on
|
1. The neonatal period (first 4 weeks of life)
2. Infancy (first year) 3. Age 1 to 4 years 4. Age 5 to 14 years |
|
|
2. Three leading causes of death during the first 12 months of life
|
1. Congenital anomalies
2. Disorders relating to gestation and low birth weight 3. SIDS |
|
|
3. What are malformations?
|
Malformations represent primary errors of morphogenesis, in other words there is an intrinsically abnormal developmental process.
They are usually multifactorial rather than the result of a single gene or chromosomal defect. |
|
|
4. What are disruptions?
|
Result from secondary destruction of an organ or body region that was previously normal in development; thus they arise from an extrinsic disturbance in morphogenesis.
Ex: amniotic bands that encircle, compress or attach to developing parts of the fetus |
|
|
5. What is the most common underlying factor responsible for deformations?
|
Uterine constraint.
|
|
|
6. What is a sequence?
What is an example? |
A sequence is a pattern of cascade anomalies. Approx 1/2 the time, congenital anomalies occur singly; in the remaining cases, multiple congenital anomalies are recognized.
A good example is the oligohydroamnios (or Potter) sequence. Oligohydramnios may be cause the infant to be compressed. This results in a classic phenotype in the newborn infant, including flattened facies and positional abnormalities of the hands and feet. Growth of the chest wall and the contained lungs is also compromised so that the lungs are freq hypoplastic. |
|
|
7. What is a syndrome?
|
A syndrome is a constellation of congenital anomalies believed to be pathologically related, that, in contrast to a sequence, cannot be explained on the basis of a single, localized, initiating defect.
Syndromes are most often caused by a single etiologic agent, such as a viral infection or specific chromosomal abnormality, which simultaneously affects several tissues. |
|
|
8. What is the difference between hyperplasia and hypertrophy?
|
Hyperplasia refers to the overdevelopment of an organ associated with increased number of cells.
An abnormality in an organ or a tissue as a result of an increase in size of individual cells defines hypertrophy. |
|
|
9. Three groups of anomalies that are genetic in origin
|
1. Those associated w/karyotypic aberration
2. Those arising from single gene mutations 3. Multifactorial inheritance |
|
|
10. What are the characteristics of chromosomal syndromes?
|
Characterized by congenital anomalies; ***the great prevalence of these cytogenic aberrations arises as defects in gametogenesis and so are not familial***
*except for a form of Down syndrome associated with a Robertsonian translocation |
|
|
11. What are single gene mutations?
|
May underlie major congenital anomalies, which as expected, follow Mendelian patterns of inheritance.
Of these, approx 90% are inherited in an autosomal dominant or recessive pattern, while the remainder segregates in an X-linked pattern. |
|
|
12. Gene involved in pts with anomalies of digits, either syndactyly or polydactyly?
|
Mutations of a downstream target of sonic hedgehog signaling, GLI3, have been reported in these pts.
|
|
|
13. Gene involved in developmental patterning and holoprosencephaly
|
Sonic hedgehog (SHH)
|
|
|
14. At risk period for rubella infection
|
The at risk period for rubella infection extends shortly before conception to the 16th week of gestation; the hazard being greater in the first 8 weeks.
Leads to rubella embryopathy |
|
|
15. What are the four main characteristics of rubella embryopathy?
|
1. Cataracts
2. Heart defects (PDA, pulmonary artery hypoplasia or stenosis, ventricular septal defect, tetralogy of Fallot) 3. Deafness 4. Mental retardation |
|
|
16. At risk period for cytomegalovirus?
What are the most prominent clinical features of CMV infection in utero? |
The highest at-risk period is the second trimester of pregnancy.
*Involvement of the CNS is a major feature, and the most prominent clinical changes are mental retardation, microcephaly, deafness, and hepatosplenomegaly. |
|
|
17. What are the characteristics of fetal alcohol syndrome?
|
1. Growth retardation
2. Microcephaly 3. ASD 4. Short palpebral fissures 5. Maxillary hypoplasia |
|
|
18. Exposure to high amts of radiation during the period of organogenesis leads to...?
|
Leads to malformations, such as microcephaly, blindness, skull defects, spina bifida, and other deformities.
|
|
|
19. What are the characteristics of babies born with diabetic embryopathy?
(Maternal hyperglycemia-induced fetal hperinsulinemia) |
Maternal hyperglycemia-induced fetal hyperinsulinemia results in increased body fat, muscle mass, and organomegaly; cardiac anomalies, neural tube defects, and other CNS malformations.
|
|
|
20. In the early embryonic period, when the the embryo most susceptible to teratogenesis?
|
Between weeks 3-9, the embryo is extremely susceptible to teratogenesis, and the peak sensitivity during this period occurs between weeks 4-5.
|
|
|
21. Teratogens and genetic defects may act at 6 steps involved in normal morphogenesis, which are:
|
1. Proper cell migration
2. Cell proliferation 3. Cellular interactions 4. Cell-matrix association 5. Programmed cell death 6. Hormonal influences and mechanical forces |
|
|
22. What is the relationship between retinoic acid and TGF and FGF?
|
TGF and FGF are both involved in morphogenesis; abnormal expression of TGF and FGF have been found in the developing palate in those animals exposed to retinoic acid.
***Thus, retinoic acid can lead to cleft palate and cleft lip.*** However, a retinoic acid derivative, all-trans-retinoic acid, is essential for normal development and its absence results in a constellation of malformations; thus the need for Vitamin A. |
|
|
23. What are HOX genes?
|
Homeobox genes; involved in transcriptional regulation
Have a 180 nucleotide motif which has DNA binding properties; these genes have been implicated in the patterning of limbs, vertebrae, and craniofacial structures. Interacts w/many other genes |
|
|
24. Mutations in HOXD13 cause...?
|
Synpolydactyly (extra digits) in heterozygous individuals
|
|
|
25. Mutations in HOXA13 cause...?
|
Hand-foot-genital-sydnrome, characterized by distal limb and distal urinary tract malformations.
|
|
|
26. What are PAX genes?
|
A 384 base pair sequence family of developmental genes.
In contrast to HOX genes, however, their expression patterns suggest they act singly. Mutations in PAX genes cause human malformations. |
|
|
27. Mutations in PAX3 cause?
|
PAX3 is mutated in Waardenburg syndrome, characterized by congenital pigment abnormalities and deafness.
|
|
|
28. Muatations in PAX6 cause...?
PAX2? |
PAX6 mutations cause congenital absence of the iris - aniridia
PAX2 mutations cause the "renal-coloboma" syndrome, characterized by developmental defects of the kidneys, eyes, ears, and brain. |
|
|
29. How are PAX genes associated with tumorigenesis?
|
PAX genes may function also as proto-oncogenes, in that their overexpression is associated with tumorigenesis.
Translocations involving PAX3 and PAX7 have been identified in the majority of alveolar rhabdomyosarcomas, and translocations involving PAX5 and PAX8 are observed in subsets of lymphomas and thyroid CAs, respectively. |
|
|
30. What is AGA?
|
Appropriate for gestational age
weight falls between the 10th and 90th percentiles |
|
|
31. What is SGA?
|
Small for gestational age
weight falls below 10th percentile Caused by fetal growth restriction (FGR) |
|
|
32. What is LGA?
|
Large for gestational age
Weight falls above 90th percentile |
|
|
33. What is the definition of prematurity?
|
Defined by a gestational age less than 37 weeks (and also weigh less than 2500 gm)
*It is the second most common cause of neonatal mortality (congenital anomalies is first). |
|
|
34. What are four risk factors for prematurity?
|
1. Preterm premature rupture of placental membranes (PPROM)
2. Intrauterine infections 3. Uterine, cervical, and placental structural abnormalities 4. Multiple gestation (twin pregnancy) |
|
|
35. What are the three most common risk factors for PPROM?
|
1. Maternal smoking
2. A prior history of preterm delivery 3. Vaginal bleeding at any time during the index pregnancy |
|
|
36. What are the most common microorganisms in intrauterine infections?
What labs would indicate intrauterine infection? |
1. Ureaplasma urealyticum
2. Mycoplasma hominis 3. Gardnerella vaginalis 4. Trichomonas 5. Gonorrhea 6. Chlamydia ***Intrauterine infections can be clinically silent or culture-negative; in these instances, elevated intra-amniotic levels of cytokines (e.g., IL-6), or maternal granulocyte colony stimulating factor are usually demonstrable. |
|
|
37. What are two ways in which intrauterine infections can initiate the onset of preterm labor?
|
1. The release of collagenases and elastases, with consequent rupture of placental membranes
2. Release of prostaglandins that induce uterine smooth muscle contractions. |
|
|
38. What are five consequences of fetal growth restriction?
|
1. Hyaline membrane disease (respiratory distress syndrome)
2. Necrotizing enterocolitis 3. Sepsis 4. Intraventricular hemorrhage 5. Long term complications, including developmental delay |
|
|
39. What is the relationship between SGA and fetal growth restriction?
What causes FGR? |
Although preterm infants have low birth weights, it is often appropriate once adjusted for their gestational age. In contrast, at least one third of infants who weigh less than 2500 gm are born at term and therefore are undergrown rather than immature.
*Hence, FGR commonly underlies SGA. Can be detected before delivery by US. Caused by: fetal factors, placental factors, and maternal factors. |
|
|
40. What are the fetal factors that reduce the growth potential?
How can you tell if infants that are SGA b/c of fetal factors? |
1. Chromosomal disorders
2. Congenital anomalies 3. Congenital infections -The TORCH group ***Infants who are SGA b/c of fetal factors are usually characterized by symmetric growth restriction, meaning that all organ systems are similarly affected |
|
|
41. TORCH
What does it mean? |
Toxoplasmosis
Other viruses, such as syphillis Rubella Cytomegalovirus Herpes virus |
|
|
42. What are five placental factors that reduce the growth potential?
How can you tell if an infant is SGA b/c of placental factors? |
1. Uteroplacental insufficiency
2. Umbilical-placental vascular anomalies 3. Placenta abruptio 4. Placenta previa 5. Confined placental mosaicism ***Placental causes of FGR tend to result in asymmetric growth retardation with relative sparing of the brain |
|
|
43. What is an important cause of growth restriction by placental factors?
|
Uteroplacental insufficiency is an important cause of growth restriction.
This insufficiency may result from umbilical-placental vascular anomalies (such as single umbilical artery, abnormal cord insertion, placental hemangioma), placenta abruptio, placenta previa, placental thrombosis and infarction, or multiple gestations. |
|
|
44. What is confined placental mosaicism?
What is the most frequent abnormality documented in confined placental mosaicism? |
Confined placental mosaicism is a cause of FGR and results from viable genetic mutations occurring after zygote formation. If it occurs early, it results in generalized constitutional mosaicism of the fetus and placenta.
Conversely, if the mutation occurs later, a genetic abnormality limited to the placenta results (confined placental mosaicism). *Chromosomal trisomies, in particular trisomy 7, are the most freq abnormality documented. |
|
|
45. What are 5 maternal factors that reduce the growth potential?
|
1. Preeclampsia
2. Chronic hypertension 3. Narcotics abuse 4. EtOH intake 5. Malnutrition |
|
|
46. What are the features of preterm lungs?
|
The alveoli are small and the septa are considerably thicker than in the adult.
Development of alveoli continues after birth and the full adult stage is reached at age 8. The immature lungs are grossly unexpanded, red, and meaty. |
|
|
47. What is the morphology of the preterm lungs?
|
*The alveolar spaces are incompletely expanded and often lined by cuboidal epithelium and usually contain pink proteinaceous precipitate and occasional squamous epithelial cells.
|
|
|
48. What is the morphology of the preterm kidneys?
|
In the preterm infant, the formation of glomeruli is incomplete. The primitive glomeruli have an organoid, glandular appearance b/c of the cuboidal cells in the parietal and visceral layers of Bownman capsule.
The deep glomeruli are well formed, however, and renal function is adequate to permit survival. |
|
|
49. Morphology of the preterm brain?
|
Incompletely developed in the preterm infant.
Brain substance is easily torn, there is poorly developed myelination of the nerve fibers. Homeostasis is not perfect, and the preterm infant has difficulty maintaining a constant body temp. |
|
|
50. Morphology of the preterm liver?
|
Suffers from a lack of physiologic maturity in the preterm infant.
Have a transient period of physiologic jaundice within the first post natal week. |
|
|
51. What is an Apgar score?
|
Method of clinically evaluating the physiologic condition and responsiveness of newborn infants.
Infant may be evaluated at 1 min and at 5 min. A total score of 10 indicates an infant in the best possible condition |
|
|
52. What 5 things does the Apgar specifically measure?
|
1. Heart rate
2. Respiratory effort 3. Muscle tone 4. Response to catheter in nostril 5. Color |
|
|
53. Apgar chart
|
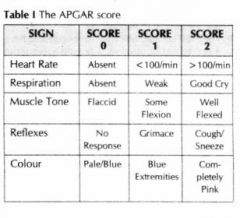
|
|
|
54. What is the most common and most important birth injury?
|
*Intracranial hemorrhages*
Generally related to excessive molding of the head or sudden pressure changes in its shape as it's subjected to the pressure of forceps or sudden precipitate expulsion. May arise from tears in the dura or from rupture of the vessels that traverse the brain. |
|
|
55. What is caput succedaneum and cephalhematoma?
|
Progressive accumulation of interstitial fluid in the soft tissues of the scalp giving rise to a usually circular area of edema, congestion, and swelling at the site where the head begins to enter the uterine canal (caput succedaneum).
Hemorrhage may occur in the scalp, producing a cephalhematoma. |
|
|
56. How are fetal and perinatal infections acquired?
(two ways) |
Through one of two routes:
1. Transcervically (ascending) 2. Transplacentally (hematologic) |
|
|
57. What are the features of transcervical infections?
|
The fetus eithers inhales infected amniotic fluid into the lungs shortly before birth or by passing through an infected birth canal during delivery.
Can cause: 1. Pneumonia 2. Sepsis 3. Meningitis |
|
|
58. What are the features of transplacental infections?
|
Most parasitic (toxoplasma, malaria) and viral/bacterial infections (listeria, Treponema) gain access to the fetal bloodstream via the chorionic villi.
|
|
|
59. What is fifth disease?
|
Caused by **parvovirus B19** which can induce spontaneous abortion, stillbirth, hydrops fetalis, and congenital anemia.
|
|
|
60. What are the similar clinical and pathologic manifestations of the TORCH group of infections?
8 of them... |
1. Encephalitis
2. Fever 3. Chorioretinitis 4. Hepatosplenomegaly 5. Pneumonitis 6. Myocarditis 7. Hemolytic anemia 8. Vesicular or hemorrhagic skin lesions |
|
|
61. What is respiratory distress syndrome (RDS)?
|
AKA hyaline membrane disease.
Occurs primarily in the immature lung. ***Caused by a deficiency of the pulmonary surfactant synthesized by type II pneumocytes.** Type II pneumocytes are most abundant after 35 wks gestation. Decreased surfactant results in increased alveolar surface tension, progressive alveolar atelectasis, and increasing inspiratory pressures required to expand alveoli |
|
|
62. What are the consequences of RDS?
|
Hypoxemia results in acidosis, pulmonary vasoconstriction, pulmonary hypoperfusion, capillary endothelial and alveolar epithelial damage, and plasma leakage into alveoli.
Plasma proteins combine w/fibrin and necrotic alveolar pneumocytes to form hyaline membranes. |
|
|
63. What is the morphology of the lungs in RDS?
|
Grossy, lungs are solid, airless, and reddish purple.
Microscopically, alveoli are poorly developed and freq collapsed. Proteinaceous membranes line respiratory bronchioles, alveolar ducts, and random alveoli. |
|
|
64. What is the clinical presentation of a neonate with RDS?
Can it be treated in utero? |
Typical infant is preterm but appropriate for gestational age.
Associated w/maternal diabetes (surfactant synthesis is suppressed by high insulin levels) and cesarean section) Can measure amniotic fluid phospholipids to assess fetal surfactant synthesis. If low, prophylactic admin of surfactant at birth to extremely premature neonates and symptomatic admin to older premature neonates is highly beneficial. |
|
|
65. What is the role of glucocorticoids in the production of surfactant?
|
Surfactant synthesis is modulated by hormones and growth factors, including cortisol, insulin, prolactin, thyroxine, and TGF-beta.
The role of glucocorticoids is important; corticosteroids induce the formation of surfactant lipids and surfactant associated proteins in fetal lung; administration of antenatal corticosteroids decrease neonatal morbidity and mortality when administered to mothers with threatened premature delivery at 24-34 weeks gestation. |
|
|
66. What is necrotizing enterocolitis (NEC)?
|
Most commonly occurs in premature infants; the incidence is inversely proportional to gestational age.
Intestinal ischemia is a prerequisite, other predisposing conditions include bacterial colonization of the gut and administration of formula feeds, both of which aggravate mucosal injury in immature bowel |
|
|
67. What is the pathogenesis of NEC?
|
Some postnatal insult (intro of bacteria) sets in motion the cascade culminating in tissue destruction.
Intestinal ischemia appears to be the prerequisite. Inflammatory mediators such as platelet-activating factor, have been implicated. |
|
|
68. Clinical course of NEC
|
Includes bloody stools, abdominal distention and development of circulatory collapse.
Gas is often present in the intestinal wall (pneumatosis intestinalis). NEC typically involves the terminal ileum, cecum, and right colon. Resection usually required. |
|
|
69. What is germinal matrix (intraventricular hemorrhage)?
|
Subependymal (germinal matrix) hemorrhage, with secondary bleeding into the ventricles, is particularly prone to occur in preterm infants who are SGA.
The microcirculation within the germinal matrix is susceptible to damage from hypoxia and changes in perfusion pressure, both relatively common occurrences in apneic preterm babies. *These cause sudden increases in intracranial pressure, damage to the brain substance, herniation of the medulla or base of brain into the foramen magnum, and serious, freq fatal depression of function of the vital medullary centers. |
|
|
70. What is immune hydrops?
|
*Defined as a hymolytic disease in the newborn caused by blood-group incompatibility between mother and child.
It occurs when the fetus inherits erythrocyte antigens from the father that the mother lacks. A small transplacental bleed allows fetal erythrocytes to enter the maternal circulation where they induce antibody production. These maternal antibodies then cross the placenta after multiple exposures and this results in erythrocyte lysis in the newborn. |
|
|
71. Although ABO incompatibility is more common than Rh incompatibility, hemolytic disease severe enough to require treatment is rare in such mismatches because...?
Three reasons... |
1. Most anti-A and anti-B antibodies are IgM and do not cross the placenta
2. Neonatal RBCs poorly express A and B blood group antigens. 3. Many cells in addition to RBCs express A and B antigens and therefore adsorb antibody that enters the fetal bloodstream. |
|
|
72. What are three major causes of nonimmune hydrops?
|
1. Cardiovascular defects
2. Chromosomal anomalies (Turner syndrome, trisomies 21 and 18) 3. Fetal anemia unrelated to immune hemolysis (e.g. β˚β˚ thalassemia, and α-thalassemia *most common) * Transplacental infection by parvovirus B19 is also rapidly emerging as an important cause of hydrops; the virus enters and replicates within erythroid precursors (normoblasts), leading to erythrocyte maturation arrest and aplastic anemia. |
|
|
73. What is the morphology of hydrops associated w/fetal anemia?
Five features... |
1. Both fetus and placenta are characteristically pale
2. The liver and spleen are enlarged from cardiac failure and congestion 3. Bone marrow demonstrates compensatory hyperplasia of erythroid precursors 4. Extramedullary hematopoiesis occurs in liver, spleen, and occasionally other tissues. 5. Increased hematopoiesis results in large numbers of immature RBCs in the peripheral blood, hence the name erythroblastosis fetalis |
|
|
74. What is the most serious threat in fetal hydrops?
|
CNS damage known as kernicterus.
The brain is enlarged and edematous and when sections is found to have a bright yellow pigmentation, particular in the basal ganglia, thalamus, cerebellum, cerebral gray matter, an spinal cord. |
|
|
75. What is phenylketonuria PKU?
|
Approx 50% of dietary phenylalanine is required for protein synthesis; the remainder is converted into tyrosine by the phenylalanine hydroxylase system (PAH).
Most freq mutation is classic PKU and is relatively common in people of Scandinavian descent and uncommon in blacks and Jews. *Homozygotes for this autosomal recessive disorder classically have a severe deficiency of PAH, leading to hyperphenylalanimeia and its pathologic consequences. |
|
|
76. What are the characteristics of PKU?
|
Although normal at birth, affected individuals exhibit rising plasma phenylalanine w/in the first few weeks of life, followed by impaired brain development and mental retardation. IQ levels are usually around 50-60.
Screening at birth for abnormally elevated urinary levels of various phenylalanine metabolites allows early diagnosis. Subsequent phenylalanine dietary restriction prevents most clinical sequelae. |
|
|
77. What is benign hyperphenylalanemia?
|
Some mutations in the PAH gene result in a partial enzyme deficiency and therefore only moderately elevated phenylalanine levels.
Such patients suffer no neurologic sequelae. |
|
|
78. What is the biochemical abrnomlity in PKU?
|
An inability to convert phenyalanine into tyrosine. Abnormalities in phenyalanine hydroxylase cause 98-99% of cases of PKU.
With a block in phenylalanine metabolism owing to lack of PAH, minor shunt pathways come into play, yielding phenylpyruvic acid, phenyllactic acid, phenylacetic acid, and o-hydroxyphenylacetic acid, which are excreted in large amts in the urine in PKU. ***There is a strong musty or mousy odor to affected infants. |
|
|
79. What is type 2 PKU?
Type 3 PKU? |
Some pts lack the enzyme responsible for BH₄ regeneration, dihydropteridine reductase (type 2 PKU).
Others lack the biopterin synthetic enzyme, dihydrobiopterin synthase (type 3 PKU). ***IT is important to recognize these PKU variants because the ongoing neurologic disturbances cannot be treated by dietary control of phenylalanine levels alone. |
|
|
80. What is galactosemia?
|
Dietary lactose, is split into glucose and galactose by lactase; galactose is then converted to glucose by three additional enzymes.
***The most common and clinically significant form of galactosemia is autosomal recessive, resulting from mutation in galactose-3-phosphate uridyl transferase (GALT).*** Affected patients accumulate galactose-1-phosphate. |
|
|
81. What is the clinical picture in galactosemia?
|
Variable, corresponding to distinct mutations in GALT.
Overall, infants fail to thrive, and present with vomiting and diarrhea after milk ingestion. Liver, eyes and brain are most severely affected; changes include hepatomegaly due to hepatic fat accumulation, followed by cirrhosis, cataracts, and nonspecific alterations in the CNS (including mental retardation). There is also and increased freq of E. coli septicemia, and hemolysis and coagulopathy in the newborn can occur as well. |
|
|
82. What is the treatment for galactosemia?
|
Urinary screening at birth reveals the presence of an abnormal reducing sugar.
Removal of dietary galactose for at least the first two years of life prevents most of the clinical and morphologic sequelae. |
|
|
83. What is the most prevalent genetic mutation in non-hispanic whites for galactosemia?
What about for African Americans? |
A glutamine to arginine substitution at codon 188 (Gln188Arg) is the most prevalent mutation in non-hispanic whites for galactosemia.
A serine to leucine substitution at codon 135 (Ser135Leu) is the most common mutation in African Americans. |
|
|
84. What is cystic fibrosis (CF)
|
Most common lethal genetic disease affecting whites.
It affects epithelial ion transport resulting in abnormal fluid secretion in exocrine glands and in respiratory, GI and reproductive mucosa. |
|
|
85. Gene responsible for CF and consequences of mutations in this gene
|
The gene responsible for CF encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein.
CFTR regulates the movement of multiple ions as well as affecting other cellular processes; however, CFTR is primarily a chloride channel, and mutations in the CFTR gene disrupt epithelial chloride transport. Thus, normal sweat ducts require CFTR for resorption of chloride; inability to resorb chloride causes increased sweat chloride concentration |
|
|
86. What is the primary defect in cystic fibrosis?
|
The primary defect in cystic fibrosis results from abnormal function of an epithelial chloride channel protein encoded by the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome ban 7q31.2.
|
|
|
87. What is CFTR?
|
Has two transmembrane domains, two nucleotide binding domains, and a regulatory domain that contains protein kinase phosphorylation sites.
Various mutations in the CF gene affect different regions of CFTR, resulting in distinct functional consequences and differing severity of clinical sequelae. |
|
|
88. Activation of the CFTR channel is mediated by what...?
|
Activation of the CFTR channel is mediated by agonist induced increases in cAMP, followed by activation of a protein kinase A.
|
|
|
89. What does CFTR regulate?
|
CFTR regulates multiple additional ion channels and cellular processes.
It not only regulates chloride-conductance channels, but it also regulates, the potassium channels, the epithelial sodium channels (ENaC), gap junction channels, and cellular processes involved in ATP transport and mucus secretion. |
|
|
90. Of all the ion channels CFTR regulates, which one has possibly the most pathophysiolgic relevance in cystic fibrosis?
|
The interaction of CFTR with ENaC.
The ENaC is situated on the apical surface of exocrine epithelial cells, and is responsible for intracytoplasmic sodium transport from the luminal fluid, rendering it (the luminal fluid) hypotonic. The ENaC is inhibited by normally functioning CFTR; hence in cystic fibrosis, ENaC activity increases, markedly augmenting sodium transport across the apical membrane. |
|
|
91. What is the exception to this increased in sodium transport across the apical membrane in CF?
|
In human sweat ducts, where ENaC activity decreases as a result of CFTR mutations; therefore, a hypertonic luminal fluid containing both high sweat chloride and high sodium content is formed.
|
|
|
92. What is CFTR's role in the respiratory and intestinal epithelium?
|
CFTR forms one of the most important avenues for active luminal secretion of chloride. At these sites, CFTR mutations result in loss or reduction of chloride secretion into the lumen.
Active luminal sodium absorption is also increased (due to loss of inhibition of ENaC activity) |
|
|
93. So, a decreased chloride secretion and increased sodium absorption at the luminal membrane leads to what in the lungs...?
|
Both of these ion changes increase passive water reabsorption from the lumen, lowering the water content of the surface fluid layer coating mucosal cells.
|
|
|
94. So what causes the pathogenesis of respiratory and intestinal complications in CF?
|
These problems appear to stem from an isotonic but low-volume surface fluid layer.
In the lungs, this dehydration leads to defective mucociliary action and the accumulation of hyperconcentrated, viscid secretions that obstruct the air passages and predispose to recurrent pulmonary infections. |
|
|
95. What does CFTR have to do with bicarbonate ions?
|
CFTR can also transport bicarbonate ions, and in some CFTR mutant variants, chloride transport is completely or substantially preserved, while bicarbonate transport is markedly abnormal.
This leads to a decrease luminal pH which can cause adverse effects such as increased mucin precipitation and plugging of ducts, and increased binding of bacteria to plugged mucins. ***Pancreatic insufficiency, a feature of classic CF, is virtually always present when there are CFTR mutations with abnormal bicarbonate conductance.*** |
|
|
96. What are the six classes of CFTR mutations?
|
Class 1. Defective protein synthesis
Class 2. Abnormal protein folding, processing, and trafficking Class 3. Defective regulation Class 4. Decreased conductance Class 5. Reduced abundance Class 6. Altered regulation of separate ion channels |
|
|
97. What is the most common mutation in CF?
|
A class 2 mutation that leads to a three-nucleotide deletion coding for phenylalanine at position F508;
This results in defective intracellular CFTR processing w/degradation before it reaches the cell surface. Patients homozygous for the F508 mutation have virtual absence of CFTR function; they present w/severe disease. This mutation can be found in approx 70% of CF patients. |
|
|
98. Which classes are usually associated with a milder phenotype of CF?
|
Classes 4 and 5
These classes have mutations on one or both alleles that results in a less severe phenotype. |
|
|
99. Which classes are usually associated with a more severe phenotype of CF?
|
Classes 1, 2, and 3 mutations produce virtual absence of membrane CFTR and are associated with the classic CF phenotype (pancreatic insufficiency, sinopulmonary infections, and GI symptoms).
|
|
|
100. Individuals that present with what unrelated clinical phenotypes may also harbor CFTR mutations?
|
1. Idiopathic chronic pancreatitis
2. Late-onset chronic pulmonary disease 3. Idiopathic bronchiectasis 4. Obstructive azoospermia caused by BAVD These patients my not demonstrate other features of CF, yet they harbor bi-allelic CFTR mutations. These subsets are classified as non-classic or atypical CF. |
|
|
101. What two other genes play a role in modifying the frequency and severity of organ-specific manifestations in CF?
|
1. A cystic fibrosis modifier locus (CFM1) which influences the incidence and severity of meconium ileus has been recently mapped to chromosome 19q13.
2. Another candidate genetic modifier is mannose-binding lectin, a key effector of innate immunity involved in opsonization and phagocytosis of microorganisms. Functional polymorphisms in one or both mannose-binding lectin alleles associated with lower circulating levels of the protein confer a 3x higher risk of end-stage lung disease and reduced survival following chronic bacterial infection in the setting of CF. |
|
|
102. What environmental modifiers might also cause significant phenotypic differences in the CFTR genotype?
|
In pulmonary disease, concurrent viral infection predispose to colonization of pseudomonas aeruginosa species.
The static mucus creates a hypoxic microenvironment in the airway surface fluid, which in turn favors the production of alginate, a mucoid polysaccharide capsule. Alginate production protects bacteria from antibodies or antibiotics, allowing them to evade host defenses and cause chronic destructive lung disease. |
|
|
103. CF and pancreas
|
Abnormalities occur in 85-90% of patients; they range from mucus accumulation in small ducts w/mild dilation to total atrophy of the exocrine pancreas, leaving only islets within fibrofatty stroma.
Absence of pancreatic exocrine secretions impairs fat absorption; resulting deficiency of vitamin A occurs |
|
|
104. CF and intestines
|
Thick viscous plugs of mucus (meconium ileus) may cause small intestinal obstruction
|
|
|
105. CF and liver
|
Plugging of bile canaliculi by mucinous material results in diffuse hepatic cirrhosis.
Hepatic steatosis is not an uncommon finding in liver biopsies. |
|
|
106. CF and salivary glands
|
Commonly involved w/progressive duct dilation, ductal squamous metaplasia, and glandular atrophy followed by fibrosis.
|
|
|
107. CF and lungs
|
**Involved in most cases and such changes are the most serious complication of CF.**
Mucus secreting cells hyperplasia and viscous secretions block and dilate bronchioles. Superimposed infections and pulmonary abscesses are common, and these give rise to chronic bronchitis and bronchiectasis. In many instances lung abscesses develop. |
|
|
108. What are the three most common organisms responsible for lung infections in CF?
|
1. Staphylococcus aureus
2. Hemophilus influenzae 3. Pseudomonas aeruginosa |
|
|
109. CF and male genital tract
|
Azoospermia and infertility occur in 95% of males surviving to adulthood, frequently w/congenital bilateral absence of the vas deferens (CBAVD).
|
|
|
110. Clinical course of CF
|
1. Pancreatic exocrine insufficiency
2. Malabsorption of fats 3. Steatorrhea 4. Fat soluble vitamin deficiencies 5. Cardiorespiratory complications -chronic cough -lung infections -COPD -cor pulmonale |
|
|
111. What is the pancreas insufficient phenotype in CF?
|
Pancreatic exocrine insufficiency occur in the majority of patients w/CF and is associated w/severe CFTR mutations on both alleles (F508/F508).
|
|
|
112. What is the pancreas sufficient phenotype in CF?
|
Those with one severe and one mild CFTR mutation (F508/R117H), or two mild CFTR mutations retain enough pancreatic exocrine function so as not to require enzyme supplementation.
The pancreas sufficient phenotype is usually not associated w/other GI complications and in general these individuals demonstrate excellent growth and development. |
|
|
113. What is the single most common cause of death (~80%) in CF patients?
|
Cardiorespiratory complications, such as persistent lung infections, obstructive pulmonary disease, and cor pulmonale.
|
|
|
114. After cardiorespiratory complications, what is the next most common cause of death in CF patients?
|
Liver disease.
Most studies suggest that symptomatic or biochemical liver disease in CF has its onset at or around puberty, w/a prevalence of 13-17%. |
|
|
115. Dx of CF
|
Sequencing of the CFTR gene is the gold standard.
The Dx can be based on elevated sweat electrolyte concentrations > 60 mM/L. Measurement of nasal transepithelial potential difference in vivo can also be useful, as individuals w/CF demonstrate significantly more negative baseline nasal potential difference than controls. |
|
|
116. Common risk factors of SIDS (9 of them)
|
1. Infant sleeping prone
2. Prematurity 3. Low birth weight 4. SIDS in a prior sibling 5. Young maternal age 6. Short intergestational interval 7. Inadequate prenatal care 8. Low SEC status 9. Maternal smoking or drug abuse |
|
|
117. Other factors in SIDS
|
Most SIDS babies have an immediate prior history of a mild respiratory tract infection.
Also, developmental immaturity of critical brain stem regions (i.g. arcuate nucleus) involved in arousal and cariorespiratory control may play a role. |
|
|
118. Four common autopsy findings in SIDS
|
1. Mutiple petechia on thymus, visceral and parietal pleura,and epicardium
2. Histologic evidence of recent infection in the upper respiratory tract. 3. CNS demonstrates astrogliosis of the brain stem and cerebellum 4. Hypoplasia of the arcuate nucleus is reported. |
|
|
119. What is heterotopia (choristoma)?
|
Represents microscopically normal cells or tissues present in abnormal locations
These cells are usually of little significance but may be clinically confused w/true neoplasms |
|
|
120. What are hamartomas?
|
Excessive (but focal) overgrowth of cells and tissues native to the organ or site in which they occur.
They may be considered a link between malformations and neoplasms. |
|
|
121. What are hemangiomas?
|
Most common tumors of infancy; most are cutaneous, particularly on the face and scalp.
They may enlarge along w/the growth of the child, but commonly spontaneously regress. They commonly produce flat to elevated irregular, red-blue masses; some of the flat larger regions are referred to as port-wine stains. Rarely become malignant |
|
|
122. What are lymphangiomas?
|
May occur on the skin but also within deeper regions of the neck, axilla, mediastinum, and retroperitoneal tissue.
They tend to increase in size and, depending on location, become clinically significant if the encroach on vital structures. Histologically, they are composed of cystic and cavernous lymphatic spaces, with variable numbers of associated lymphocytes. |
|
|
123. What are fibrous tumors?
|
Histologically, fibrous tumors range from sparsely cellular proliferations (fibromatosis) to richly cellular lesions indistinguishable from adult fibrosarcomas.
In contrast to fibrous tumors occurring in adults, the histologic picture does not predict the biology and an individual tumor (they may spontaneously regress) |
|
|
124. What are the two peaks in teratoma incidence?
|
Teratoma incidence has two peaks: at age 2, and again in late adolescence.
Those occurring in childhood and infancy tend to occur in the sacrococcygeal region. |
|
|
125. What are sacrococcygeal teratomas?
|
Approx 10% of sacrococcygeal teratomas are associated w/congenital anomalies, primarily defects of the hindgut and cloacal region and other midline defects.
They are histologically similar to other teratomas; mesodermal, endodermal, and ectodermal elements are present. Approx 75% contain mature tissues only and are benign. |
|
|
126. In what three ways do childhood malignancies differ biologically and histologically from their adult counterparts?
|
1. A close relationship between abnormal development (teratogenesis) and tumor induction (oncogenesis)
2. A greater prevalence of underlying familial or genetic germ line aberrations 3. A tendency for some malignancies in the fetal or neonatal period to regress spontaneously or cytodifferentiate. |
|
|
127. What are the most frequent childhood cancers?
|
Those that arise in the hematopoietic system (leukemia, some lymphomas), CNS, adrenal medulla, retina, soft tissue, bone and kidney.
Leukemia accounts for more deaths in children younger than 15 years of age than all other tumors combined |
|
|
128. What are neuroblastic tumors?
|
Most neuroblastic tumors occur in children younger than 5, and arise in the adrenal medulla or various sympathetic ganglia.
Most common subtype is neuroblastoma, characterized by sheets of small, round blue neuroblasts within a neurofibrillary background and chacteristic Homer-Write pseudorosettes. Approx 90% of neuroblastomas produce catecholamines; elevated blood or urine catecholamine metabolites are important diagnostic features. |
|
|
129. What is the morphology of neuroblastomas?
|
Most common locations is the adrenal medulla. The remainder occur anywhere along the sympathetic chain. On transection, they are composed of soft, gray-tan, brainlike tissue. Histologically, they are composed of small primitive appearing cells w/dark nuclei, scant cytoplasm, and poorly defined cell borders growing in sodium sheets.
***Typically, rosettes (Homer-Write pseudorosettes) can be found.*** Immunochemical reactions may produce characteristic central dense cores surrounded by a peripheral halo (dense core granules). |
|
|
130. What is a ganglioneuroblastoma?
|
Larger cells having m ore abundant cytoplasm w/large vesicular nuclei and a prominent nucleolus, representing ganglion cells in various stages of maturation, may be found in tumors admixed with primitive neuroblasts (ganglioneuroblastoma).
|
|
|
131. What is a ganglioneuroma?
|
Even better-differentiated lesions contain many more large cells resembling mature ganglion cells w/no or minimal residual neuroblasts; such neoplasms are called ganglioneuromas.
|
|
|
132. What is a histologic prerequisite for the designation of ganglioneuroblastoma and ganglioneuroma?
Why is it important? |
The presence of a so-called "schwannian stroma" comprised of organized fascicles of neuritic processes, mature Schwann cells, and fibroblast is a histologic prerequisite.
***Documenting the presence of schwannian stroma is essential b/c it is associated with a favorable histology. |
|
|
133. What are the most important factors in childhood neoplasms?
|
Age and stage
Infants (<12 months) have an excellent prognosis regardless of stage, but children older than 5 usually have extremely poor outcomes. |
|
|
134. Six favorable/unfavorable factors important in prognosis of childhood neoplasms
|
1. Histological features (schwannian stroma is favorable)
2. Tumor ploidy (diplody, near-diploidy, or near-tetraploidy are unfavorable 3. N-myc amplification (unfavorable) 4. 17q gain and 1p loss (unfavorable) 5. Telomerase overexpression (unfavorable) 6. TrkA expression (favorable) |
|
|
135. What is a Wilms tumor?
What genetic mutation is associated with these tumors? |
Wilms tumor of the kidney is usually Dx'ed between 2 and 5 y/o. Most common primary renal tumor of childhood and the 4th most common pediatric malignancy in the uS.
Although malignant, the overall survival rate is > 90%. Although 90% are sporadic, there are associations with three groups of malformation syndromes, all involving chromosome 11p |
|
|
136. Three groups of malformation syndromes, all involving chromosome 11p
Very important card!!! |
1. WAGR (Wilms tumor, aniridia, genital anomalies, mental retardation). Patients have a 33% chance of developing Wilms tumors. WAGR involves deletion on chromosome 11p band 12
2. Denys-Drash syndrome; patients have gonadal dysgenesis and nephropathy leading to renal failure (***diffuse mesangial sclerosis); most develop Wilms tumors; dominant mutation is a dominant negative missense mutations in the zinc-finger region of WT1 gene. 3. Beckwith-Wiedemann syndrome -Patients have enlarge body organs, hemihypertrophy, renal medullary cysts, adrenal cytomegaly and a predisposition to developing Wilms and other primitive tumors (Chromosome 11p15.5) |
|
|
137. What are nephrogenic rests?
|
Nephrogenic rests are putative precursor lesions of Wilms tumors and are seen in the renal parenchyma adjacent to approx 40% of unilateral tumors; this freq rises to nearly 100% in cases of bilateral Wilmns tumors.
The appears varies from expansile masses that resemble Wilms tumors to sclerotic rests consisting predominantly of fibrous tissue and occasional admixed immature tubules or glomeruli. *These pts are at an increased risk of developing Wilms tumors in the contralateral kidney and require freq and regular surveillance for many years. |
|
|
138. What is the morphology of WIlms tumors?
|
They are large, solitary, well circumscribed renal masses.
On cut section, the tumor is soft, homogeneous, and tan to gray w/occasional focal of hemorrhage, cyst formation and necrosis. |
|
|
139. What are the three micrscopic features of Wilms tumors?
(i.e. what is the classic triphasic combination?) |
Characterized by triphasic histological features:
1. Blastema 2. Immature stroma 3. Tubules - an attempt to recapitulate nephrogenesis |
|
|
140. What are the clinical features of Wilms tumors?
What type of tumors have the least favorable outcome? |
Most children w/Wilms tumors present w/a large abdominal mass that may be unilatera or, when very large, may extend across the midline and down into the pelvis.
Hematuria, pain in the abdomen after some traumatic incident, interstinal obstruction, and appearance of hypertension ar other patterns of presentation. In a considerable number of these pts, pulmonary metastases are present at the time of primary Dx. *Tumors w/diffuse anaplasia, especially those with extrarenal spread, have the least favorable outcome. |
|
|
141. What are the three principal ways in which intertissue regulation for metabolic homeostasis is achieved?
|
1. The concentration of nutrients or metabolites in the blood affects the rate at which they are used or stored in different tissues
2. Hormones carry messages to their individual target tissues about the physiologic state of the body and the current level of nutrient supply or demand 3. The CNS uses neural signals to control tissue metabolism, either directly or thru the release of hormones. |
|
|
142. Where does insulin act?
Where does glucagon act? |
Insulin promotes the storage of glucose as glycogen in liver and muscle, conversion of glucose to TAGs in liver and their storage in adipose tissue, and AA uptake and protein synthesis in skeletal muscle.
Glucagon's site of action are principally the liver and adipose tissue; it has no influence on skeletal muscle metabolism b/c muscle cells lack glucagon receptors. |
|
|
143. What dictates the release of insulin from the β-cells?
|
The release of insulin from the β-cells of the pancreas is dictated primarily by the level of glucose in the blood bathing the β-cells in the islets of Langerhans.
Increased blood glucose stimulates insulin release. |
|
|
144. What dictates the release of glucagon from the α-cells of the pancreas?
|
The release of glucagon from the α-cells of the pancreas is controlled principally thru a reduction in glucose and/or a rise in the concentration of insulin in the blood bathing the α-cells in the pancreas.
|
|
|
145. What are the main differences between insulin/glucagon and the release of other hormones of metabolic homeostasis?
Rising levels of the insulin counterregulatory hormones in the blood reflect what...? |
Of all these hormones, only insulin and glucagon are synthesized and released in direct response to changing levels of fuels in the blood.
The release of cortisol, epinephrine, and norepinephrine is mediated by neuronal signals. Rising levels of the insulin counterregulatory hormones in the blood reflect a current increase in the demand for fuel. |
|
|
145. What is the composition of insulin?
|
Insulin is a polypeptide hormone. The active form is composed to 2 polypeptide chains (the A and B-chains) linked by two interchain disulfide bonds. The A chain has an additional intrachain disulfide bond.
|
|
|
146. How is insulin synthesized?
|
Insulin is synthesized as a preprohormone that is converted in the rER to proinsulin.
Proinsulin folds into the proper conformation, and disulfide bonds are formed between the cysteine residues. It is then transported in microvesicles to the Golgi complex. It leaves the Golgi in storage vesicles, where a protease removes the biologically inactive "connecting peptide" (C-peptide), resulting in the formation of biologically active insuline. |
|
|
147. Cleavage of the C-peptide does what?
|
Cleavage of the C-peptide decreases the solubility of the resulting insulin, which then co-precipitates w/zinc.
|
|
|
148. How does glucose enter the β-cell?
|
Glucose enters the β-cell via specific glucose transporter proteins known as GLUT 2.
Glucose is phosphorylated via glucokinase to form G6P, which is metabolized thru glycolysis, the TCA cycle, and oxidative phosphorylation. These reactions result in an increase in ATP levels within the β-cell. |
|
|
149. What happens as the β-cell ATP/ADP ratio increases?
|
The activity of a membrane-bound, ATP-dependent potassium channel is inhibited (closed).
The closing of this channel leads to a membrane depolarization which activates calcium channels to increase the concentration of intracellular calcium levels in the β-cell. |
|
|
150. A rise in the intracellular calcium concentration in the β-cell has what effect?
|
This increase calcium stimulates the fusion of insulin containing exocytotic vesicles w/the plasma membrane, resulting in insulin secretion.
|
|
|
151. What is the threshold for insulin release?
|
The threshold ofr insulin release is approx 80 mg glucose/dL. Above 80, the rte o insulin release is not an all-or-nothing response but is proportional to the glucose concentration up to 300 mg/dL.
|
|
|
152. What factors other than the blood glucose concentration can modulate insulin release?
|
1. Amino acids increase insulin release
2. GIP and GLP-1, gut hormones increase insulin release 3. Epinephrine decreases insulin release |
|
|
153. How is glucagon synthesized?
|
Glucagon, a polypeptide hormone, is synthesized in the α-cells of the pancreas by cleavage of the much larger preproglucagon, a 160-AA peptide.
Like insulin, preproglucagon is produced on the rER and is converted to proglucagon as it enters the lumen of the ER. Proteolytic cleavage at various sites produces the mature 29-AA glucagon and larger glucagon-containing fragments. |
|
|
154. How glucagon secretion regulated?
|
Via circulating levels of glucose and insulin. Increasing levels of each inhibit glucagon release.
Glucose probably has both a direct suppressive effect on secretion of glucagon from the α-cell as well as an indirect effect. The direction of blood flow in the islets of the pancreas carries insulin from the β-cells in the center of the islets to the peripheral α-cells, where it suppresses glucagon secretion. |
|
|
155. What other hormones regulate glucagon secretion?
|
Amino acids increase secretion of glucagon.
Cortisol, neural stress, and epinephrine especially stimulate glucagon secretion. |
|
|
156. Why does it make sense for amino acids to stimulate insulin and glucagon secretion after a meal?
|
Insulin release stimulates AA uptake by tissues and enhances protein synthesis. However, b/c glucagon levels also increase in resposne to a protein meal, and the critical fact is the insulin:glucagon ratio, sufficient glucagon is released that gluconeogenesis is enhanced (at the expense of protein synthesis), and the AAs that are taken up by the tissues serve as a substrate for gluconeogenesis.
|
|
|
157. How do polypeptide hormones initiate their actions?
|
By binding to specific receptors or binding proteins. Insulin, glucagon, and catecholamines act thru binding to a specific receptor on the plasma membrane. The first message of the hormone is transmitted to intracellular enzymes by the activated receptor and an intracellular second messenger; no need for it to enter the cell.
|
|
|
158. How do steroid hormones act?
|
Steroid hormones such as cortisol and the thyroid hormone T3 enter the cytosol and eventually move into the cell nucleus to exert their effects.
|
|
|
159. What are the three basic types of signal transduction for hormones binding to receptors on the plasma membrane?
|
1. Receptor coupling to adenylate cyclase, which produces cAMP
2. Receptor kinase activity 3. Receptor coupling to hydrolysis of PIP₂ |
|
|
160. What is the composition of the insulin receptor?
|
The insulin receptor has two subunits, an α-subunit to which insulin binds, and the β-subunits, which span the membrane and protrude into the cytosol.
The cytosolic portion of the β-subunit has tyrosine kinase activity. |
|
|
161. How does insulin transduce its signal?
|
It binds to a receptor on the plasma membrane of its target cells.
On binding of insulin, the tyrosine kinase phosphorylates tyrosine residues on the β-subunit as well as several other enzymes w/in the cytosol. A principal substrate for phosphorylation by the receptor, insulin-receptor substrate 1 (IRS-1), then recognizes and binds to various signal transduction proteins in regions referred to as SH2 domains. |
|
|
162. What are the five major categories of the basic tissue-specific cellular responses to insulin?
|
1. Insulin reverse glucagon-stimulated phosphorylation
2. Insulin works thru a phosphorylation cascade that stimulates the phosphorylation of several enzymes 3. Insulin induces and represses the synthesis of specific enzymes 4. Insulin acts as a growth factor and has a general stimulatory effect on protein synthesis. 5. Insulin stimulates glucose and AA transport into cells. |
|
|
163. How does glucagon transduce its signal?
|
Its glucagon receptor is coupeld to adenylate cyclase and cAMP production.
Glucagon, thru G proteins, activates the bound adenylate cyclase, increasing synthesis of cAMP. cAMP activates protein kinase A, which changes the activity of enzymes by phosphorylating them at specific serine residues. |
|
|
164. What hormones inhibit adenylate cyclase?
What relfects the amt of cAMP present at any time? |
The inhibitory G-protein complex called Gi-complex.
The amt of cAMP present at any time is a direct reflection of hormone binding and the activity of adenylate cyclase. It is not affected by ATP, ADP, or AMP levels in the cells. |
|
|
165. How does cAMP affect protein kinase A?
|
As cAMP binds to the regulatory subunits of protein kinase A, these subunits dissociate from the catalytic subunits, which are thereby activated.
Activated protein kinase A phosphorylates serine residues of key regulatory enzymes in the pathways of carb and fat metabolism. Some enzymes are activated and other are inhibited by this change in phosphorylation state. |
|
|
166. What are CREs and CREBs?
What are their roles in transcription? |
Changes in the phosphorylation state of proteins that bind to cAMP response elements (CREs) in the promoter region of genes contribute to the regulation of gene transcription by a number of cAMP-coupled hormones.
For instance, cAMP response element binding protein (CREB) is directly phosphorylated by protein kinase A, a step essential for the initiation of transcription. |
|
|
167. How do the β-adrenergic receptors work when activated?
|
The three β-adrenergic receptors work thru the adenylate cylcase-cAMP system, activating a Gs-protein, which activates adenylate cyclase, and eventually protein kinase A.
|
|
|
168. What is the role of β1 receptors?
|
THe β1 receptor is the major adrenergic receptor in the human heart and is primarily stimulated by norepinephrine.
On activation, the β1 receptor increases the rate of muscle contraction, in part b/c of PKA-mediate phosphorylation of phospholamban. |
|
|
169. What is the role of the β2 receptor?
|
The β2 receptor is present in liver, skeletal muscle, and other tissues and is involved int eh mobilization of flues. It also mediates vascular, bronchial, and uterine smooth muscle contraction.
*Epinephrine is a much more potent agonist for this receptor than norepinephrine. |
|
|
170. What is the role of the α3 receptor?
|
The α3 receptor receptor is found predominantly in adipose tissue and to a lesser extent in skeletal muscle.
Activation of this receptor stimulates fatty acid oxidation and thermogenesis, and agonists for this receptor may prove beneficial in weight loss. |
|
|
171. What is one of the important cellular repsonses to insulin?
|
The reversal of glucagon-stimulated phosphorylation of enzymes.
Mechanisms of this may be inhibition of adenylate cyclase, reduction of cAMP levels, stimulation of phosphodiesterase, the production oa specific protein, the release of a second messenger, and the phosphorylation of enzymes at a site that antagonizes protein kinase A phosphorylation. |
|
|
172. How is the rate of synthesis of mRNA for PEP carboxykinase regulated?
|
This key enzyme of the gluconeogenic pathway is increased several fold by glucagon via cAMP and decreased by insulin.
This antagonism is exerted thru an insulin-sensitive hormone response element in the promoter region of the genes. |
|
|
173. How does insulin stimulate protein synthesis?
|
It increases the rates of mRNA translation for a broad spectrum of structural proteins.
These actions result from a phosphorylation cascade initiated by the autophosphorylation of the insulin receptor and ending in the phosphorylation of subunits of proteins that bind to an inhibit eukaryotic protein synthesis initiation factors (eIFs). *When phosphorylated, the inhibitory proteins are released from the eIFs, allowing translation of mRNA to be stimulated. |
|
|
174. Why does hyperglycemia cause polyuria and subsequent polydipsia?
|
These symptoms are caused by the osmotic diuresis that results from the high levels of glucose being excreted into the urine. This diuretic effect may decrease the plasma volume, thereby further increasing blood glucose levels.
|
|
|
175. What is a nonketotic hyperosmolar coma?
|
Extremely high levels of serum glucose can cause nonketotic hyperosmolar coma in pts w/type 2 DM.
Such pts usually have sufficient insulin responsiveness to block fatty acid release and ketone body formation, but they are unable to significantly stimulate glucose entry into peripheral tissues. The severely elevated levels of glucose in the blood compared with those inside the cell leads to an osmotic effect that causes water to leave the cells and enter the blood. |
|
|
176. How can on demonstrate autonomous hypersecretion of insulin from a suspected β-cell insulinoma?
|
Draw blood for the measurement of both glucose and insulin simultaneously, at a time when the pt is spontaneously experiencing the characteristic adrenergic or neuroglycopenic symptoms of hypoglcemia
During such a test, a person with an insulinoma will have glucose levels far below normal and the ration of insulin:glucose will be higher than normal. |
|
|
177. What confers susceptibility to type 1 DM?
|
Conferred by a genetic defect in the HLA region of β-cells that codes for the MHC II.
This protein presents an intracellular antigen to the cell surface for "self-recognition" by the cells. B/c of this defective protein, a cell mediated immune response destroys the β-cells. |
|
|
178. What causes MODY?
|
MODY results from specific mutations in either pancreatic glucokinase or specific nuclear transcription factors.
|
|
|
179. What causes MODY type 2?
|
MODY type 2 is due to a glucokinase mutation that results in an enzyme with reduced activity.
Individuals with this glucokinase mutation cannot significant metabolize glucose unless glucose levels are higher than normal. Therefore, they are almost always in a hyperglycemic state. |
|
|
180. What is the most common mutation leading to permanent neonatal diabetes?
|
KCNJ11 gene, which encodes a subunit of the potassium-ATP channel. This is an activating mutation, which keeps the channel open, and less susceptible to ATP inhibition.
The the channel cannot be closed, activation of the calcium channel will not occur, and insulin secretion will be impaired. |
|
|
181. How do sulfonylureas work?
|
They act on the potassium-ATP channels to close them, which, in turn, increases calcium movement into the interior of the β-cell.
This influx of calcium modulates the interaction of the insulin storage vesicles w/the plasma membrane of the β-cell, resulting in the release of insulin into the circulation. |
|
|
182. Measurement of proinsulin and C-peptide in the blood provide what type of info?
|
These can provide confirmation of an insulinoma.
Insulin and C-peptide are secreted in approx equal proportions from the β-cell, but C0peptide is not cleared from the blood as rapidly as insulin. Therefore, it provides a reasonably accurate estimate of the rate of insulin secretion. *Also useful for estimating degree of endogenous insulin secretion in diabetics, b/c exogenous insulin lacks the C-peptide. |
|
|
183. What are the levels of glucagon in diabetics?
|
Despite the presence of hyperglycemia, glucagon levels in such pts initially remain elevated, either b/c of the absence of insulin's suppressive effect or b/c of the resistance of the α-cells to insulin's suppressive effect event in the face of adequate insulin levels in type 2 pts.
Thus, diabetes may be a "bi-hormonal" disorder. |
|
|
184. Phosphodiesterase is inhibited by methylxanthines, a class of compounds that includes caffeine.
Would the effect of a methyxanthine on fuel metabolism be similar to fasting or to a high carb meal? |
Inhibition of phosphodiesterase by methylxanthine would increase cAMP and have the same effects on fuel metabolism as would an increase of glucagon and epinephrine, as in the fasted state.
Increased fuel mobilization would occur thru glycogenolysis and thru lipolysis. |