Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
138 Cards in this Set
- Front
- Back
|
What four characteristics do Myelodysplastic syndromes (MDS) show?
|
1. Ineffective hematopoiesis
2. Dysplastic morphology of cells 3. Increased apoptosis of cells 4. Peripheral blood cytopenia |
|
|
What cytogenetic changes are commonly seen in MDS?
|
1. Monosomy of 5 and 7
2. Deletion of 5q, 7q, and 20q 3. Trisomy 8 |
|
|
What two ways may MDS typically occur?
|
Either idiopathically (over age 50) or after therapy with radiation/genotoxic drugs.
|
|
|
What is characteristic of MDS changes in RBCs?
|
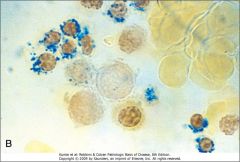
Ringed sideroblasts
|
|
|
What is characteristic of granulocytes in MDS?
|
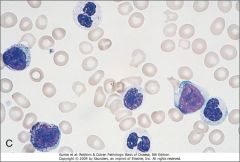
Pseudo-Pelger-Huet cells
|
|
|
What characteristic of Megakaryocytes is seen in MDS?
|
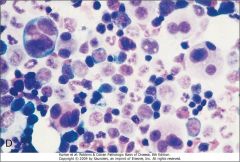
Pawn-Ball Megakaryocytes (multiple separate nuclei)
|
|
|
What is primary myelofibrosis?
|
A chronic myeloproliferative disorder characterized by obliterative marrow fibrosis.
|
|
|
What genetics are most commonly associated with primary myelofibrosis.
|
JAK-2 point mutations (50-60%).
|
|
|
What are the two stages of Primary Myelofibrosis?
|
1. Cellular stage (hypercellular).
2. Fibrotic stage (dry tap). |
|
|
What important findings would you expect for Primary Myelofibrosis?
|
1. Dry tap of bone marrow
2. Pancytopenia 3. Leukoerythroblastosis 4. Extramedullary hematopoesis 5. Tear drop cells |
|
|
What clinical findings support the diagnosis of Polycythemia vera?
|
1. ↑ erythroid precursors
2. ↑ RBC number 3. ↓ EPO 4. JAK-2 mutation (>95%) |
|
|
Broadly speaking, what genetic abnormality is associated with myeloproliferative diseases?
|
Mutations which cause constitutive activation of tyrosine kinases.
|
|
|
When evaluating anemias, what are the three broad categories you should consider?
|
1. Blood loss
2. Increased RBC destruction 3. Decreased RBC production |
|
|
What is aplastic anemia?
|
A syndrome of chronic primary hematopoietic failure with attendant pancytopenia (anemia, neutropenia, throbocytopenia).
|
|
|
Analogous to primary myelofibrosis, what is commonly seen in aplastic anemias?
|
A dry tap of the bone marrow.
|
|
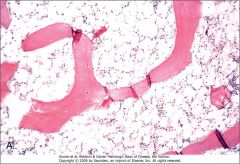
If this patient presents with pancytopenia, what is a potential diagnosis?
|
Aplastic Anemia
|
|
|
Approximately what percentage of aplastic anemia cases are idiopathic?
|
About 65% are idiopathic and the rest are spread among many different causes.
|
|
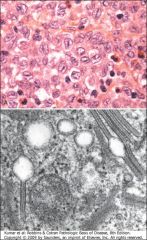
What is the diagnosis and what is the name of the structures in the lower panel?
|
Langerhans Histiocytosis. Birbeck granules.
|
|
|
What types of cells are clonally expanded in Langerhans cell histiocytosis?
|
Macrophages and dendritic cells.
|
|
|
What phrase is pathognomonic for multiple myeloma?
|
Lytic bone lesions.
|
|
|
What four clinical findings are associated with multiple myeloma?
|
1. Normocytic anemia
2. Pancytopenia 3. Lytic bone lesions 4. Bence Jones proteinuria |
|
|
In multiple myeloma, what will a serum protein electrophoresis typically show?
|
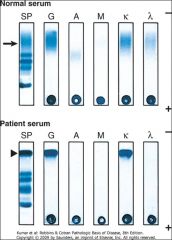
Instead of a broad, diffuse polyclonal IgG band, a sharp monoclonal IgG kappa band may be seen. This could also present as other antibody classes and it will only show elevated kappa or gamma chains.
|
|
|
What is the key cell type that you would expect to see in multiple myeloma and what is the expected immunophenotype?
|
Plasma cells (CD56, CD79a)
|
|
|
What is Bence Jones protein and what is it diagnostic of?
|
Immunoglobulin light chains (kappa or lamda). Found in multiple myeloma or Waldenstrom's macroblobulinemia.
|
|
|
What does mantle cell lymphoma look like?
|
Tumor cells which closely resemble the normal "mantle zone" cells that surround follicular centers of lymphoid tissue.
|
|
|
What is the cytogenetics of mantle cell lymphoma?
|
t(11:14) resulting in fusion of IgH and BCL 1
|
|
|
What is the immunophenotype of mantle cell lymphoma?
|
Cyclin D1, CD19, CD20, CD5
|
|
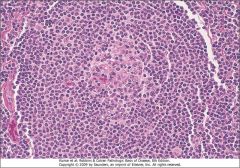
The genotype is t(11:14) and the cells are CD 5 positive, what is the diagnosis?
|
Mantle cell lymphoma.
|
|
|
What is the immunophenotype of Burkitt lymphoma?
|
CD 19, CD 20, CD 10, BCL 6
|
|
|
What is the cytogenetics associated with Burkitt Lymphoma?
|
t(8:14) c-myc/IgH
|
|
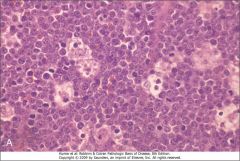
These tumor cells have a t(8:14) genotype. What is the diagnosis?
|
This is a typical starry sky pattern of dark lymphocytes and tingible body macrophages which is characteristic of Burkitt Lymphoma.
|
|
|
What do cells in marginal zone lymphomas (maltomas) resemble?
|
Normal marginal zone (memory) B cells.
|
|
|
What is an important clinical feature of marginal zone lymphomas?
|
The arise at extranodal sites that have continuous inflammation (colonic mucosa, gastric mucosa, salivary gland).
|
|
|
What are the three diseases or syndromes which predispose to extranodal marginal cell lymphomas?
|
1. Hashimoto's disease
2. Sjogren's disease 3. Helicobacter gastritis |
|
|
What is a distinguishing immunophenotype for marginal zone lymphoma?
|
CD5 and CD10 are both negative
|
|
|
What genotypes may be present?
|
t(1;14) or t(11;18)
|
|
|
What do follicular lymphoma cells resemble?
|
Normal germinal center B cells.
|
|
|
What are the two principal cell types seen in follicular lymphoma?
|
1. Small cells with irregular or cleaved nuclei (centrocytes)
2. Centroblasts with open chromatin and several nucleoli |
|
|
What is the immunophenotype of follicular lymphoma cells?
|
CD19, CD20, CD10
|
|
|
What is the genotype of follicular lymphoma cells?
|
t(14;18) IgH/BCL2
|
|
|
What is the most common form of adult lymphoma's?
|
Follicular lymphoma
|
|
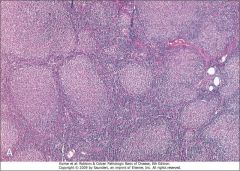
The genotype is t(14;18) and no CD5 is expressed on the tumor cells. What is the diagnosis?
|
Follicular Lymphoma
|
|
|
What is seen in the bone marrow of 85% of follicular lymphoma patients?
|
Para-trabecular lymphoid aggregates.
|
|
|
What is the major difference between SLL and CLL?
|
The degree of peripheral blood lymphocytosis. CLL, lymphocytes>4000/mm^3 in blood.
|
|
|
In SLL/CLL, what typically happens to the lymphoid architecture?
|
The nodal architecture becomes effaced.
|
|
|
What is the immunophenotype of SLL/CLL cells?
|
CD19, CD20, CD23, CD5
|
|
|
What two lymphoid disorders have cells that express CD5?
|
SLL/CLL and Mantle cell lymphoma
|
|
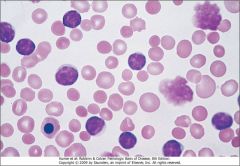
Cells were found that were CD19, CD20, CD23, and CD5 positive. What is the important clue from this image and what is the diagnosis?
|
Smudge cells indicating SLL or CLL, depending on the blood lymphocyte count.
|
|
|
Is the genotype important for diagnosing SLL/CLL?
|
No, not really.
|
|
|
What is pathognomonic for SLL/CLL?
|
Tumor with loose aggregates of prolymphocytes called proliferation centers.
|
|
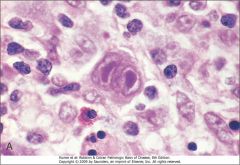
What is the diagnosis?
|
Hodgkin Lymphoma (Reed-Sternberg cell)
|
|
|
What is the immunophenotype for Hodgkin lymphoma?
|
CD20, CD45, BCL6
|
|
|
What is significant about EBV in Hodgkin lymphoma?
|
EBV is present in 40% of HLs and is important for activating NF-kB which contributes to the development of HL.
|
|
|
Are there any cytogenetics for diffuse large B-cell lymphoma?
|
No, it is quite heterogeneous.
|
|
|
What is the immunophenotype of diffuse large B-cell lymphoma?
|
CD19 and CD20; CD10 and BCL6 variable (Big Robbins)
|
|
|
Two subtypes of diffuse large B-cell lymphoma are associated with viral infections. What are these?
|
1. Immunodeficiency associated type presenting with EBV infection.
2. Primary effusion lymphoma associated with HHV-8. |
|
|
What cell type is oncogenic within a thymoma?
|
Thymic epithelial cells.
|
|
|
How are thymomas typically identified in the clinic.
|
They impinge upon mediastinal structures, or they are found in myasthenia gravis patients (autoimmune connection).
|
|
|
What is critical to remember about malignant thymoma relative to T-cell acute lymphoblastic lymphoma?
|
The only oncogenic cells are thymic epithelial cells.
|
|
|
Where does T-cell ALL appear?
|
As a thymic mass in young adults.
|
|
|
What mutations are associated with T-cell ALL?
|
Notch1
|
|
|
What is the immunophenotype of B-cell ALL?
|
CD10 CD19, TdT
|
|
|
What is a common immunophenotype between T-call and B-cell ALL?
|
TdT
|
|
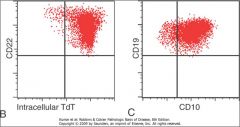
What is the likely diagnosis based on the immunophenotype?
|
B-cell ALL
|
|
|
What is the critical pathogenic event in Chronic Myeloid Leukemia (CML)?
|
Acquisition of the Philadelphia chromosome (BCR-ABL fusion gene).
|
|
|
What is the diagnostic criterion for CML?
|
Cytogenetics showing a reciprocal translocation: t(9;22) (q34;q11)
|
|
|
What is LAP and why is it important?
|
Leukocyte alkaline phosphatase is present in the granules of normal granulocytes, but absent in the granules of CML.
|
|
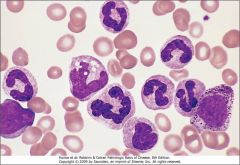
These granulocytes are LAP negative, what is the presumptive diagnosis and what test would you like to perform next?
|
Chronic Myeloid Leukemia. Cytogentic staining to look for the Philadelphia chromosome.
|
|
|
What cell types are overproduced in CML?
|
Granulocytes and megakaryocytes.
|
|
|
What are auer rods?
|
Red staining peroxidase positive azurophilic granules.
|
|
|
What cells have auer rods?
|
Myeloblasts.
|
|
|
How can you distinguish between CML and AML on peripheral blood smears?
|
Look for LAP negative leukocytes (CML) and peroxidase positive myeloblasts (AML).
|
|
|
Describe the bone marrow appearance and sites of hematopoesis in CML.
|
Bone marrow is hypercellular (~100%) and most of the erythropoeisis is extramedullary.
|
|
|
What stain is used for monoblasts?
|
non-specific esterase.
|
|
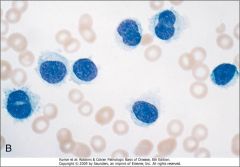
What staining procedure would you use to establish a diagnosis for this disease?
|
Tartrate resistant acid phosphatase (TRAP). If positive, then probably hairy cell leukemia.
|
|
|
What is a major difference between Waldenstrom's macroglobulinemia and multiple myeloma in terms of antibody production.
|
In MM, light chains are over-produced leading to Bence-Jones proteinuria, where as in WM, the production is balanced and renal failure/amyloidosis are rare.
|
|
|
What may cause immune hemolytic anemia with cold agglutinins?
|
Waldenstrom's macroglobulinemia because it causes overproduction of IgE which may cause autoimmune destruction of RBCs.
|
|
|
Apart from the ABO antigens, what are four additional antigens which may cause sensitization if present on transfused RBCs?
|
1. D (Rh)
2. Kell 3. Duffy 4. Kidd |
|
|
What may cause hereditary spherocytosis?
|
Mutations effecting RBC membrane integrity including: ankyrin, spectrin, band 3 or band 4.
|
|
|
What happens to RBCs in hereditary spherocytosis?
|
They age much quicker, giving off small membrane fragments and are then trapped in the spleen and eaten by macrophages.
|
|
|
How may hereditary spherocytosis be treated?
|
Splenectomy.
|
|
|
What is the critical role of G-6-P dehydrogenase in RBCs?
|
By oxidizing glucose, it provides reducing equivalents to glutathione which can protect against oxidative damage.
|
|
|
What is the average lifetime of an RBC?
|
120 days
|
|
|
What food should people suffering from G-6-PD deficiency not eat and why is this significant?
|
Fava beans. This food is endemic to the middle east which is where a large number of G-6-PD variants occur.
|
|
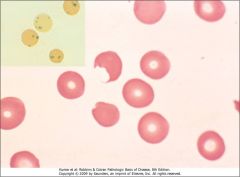
This is a blood smear stained with H&E or crystal violet from a patient with G6PD deficiency. Identify two abnormalities and how they arise.
|
1. Heinz bodies are precipitated globulins.
2. Bite cells result from macrophages plucking out these inclusions. |
|
|
Why is the penetrance of G6PD and sickle cell alleles so high in Africa?
|
Because of the heterozygote advantage against falciparum malaria.
|
|
|
What is the change in sickle cell disease?
|
Point mutation of glutamate to valine.
|
|
|
What is vaso-occlusive crisis in sickle cell disease?
|
When sickled RBCs block capilaries and lead to painful infarcts which may include organs.
|
|
|
What is autosplenectomy?
|
Over time, sickle cell patients will autoinfarct their spleen due to continuous vaso-occlusive crises.
|
|
|
Infection by what virus may lead to aplastic crisis in sickle cell patients?
|
Parvovirus B19
|
|
|
List viruses which may cause tumors and the associated tumors.
|
1. EBV: Diffuse large B-cell lymphoma (Immunodeficiency type)
2. HHV8: Diffuse large B-cell lymphoma (Body cavity tumor) 3. Adenovirus: ALL 4. HTLV-1: Adult T-cell leukemia/lymphoma |
|
|
What are the morphological classifications of anemias?
|
1. Microcytic anemias (MCV < 80 fL)
2. Normocytic anemias (80 < MCV < 100 fL) 3. Macrocytic anemias (MCV > 100 fL) |
|
|
List the four microcytic anemias.
|
1. Iron deficiency
2. Anemia of chronic disease 3. Thalassemia 4. Sideroblastic anemias |
|
|
What are macrocytic, megaloblastic anemias typically associated with?
|
Vitamin B12 and/or folate deficiency
|
|
|
How is iron abosrption regulated?
|
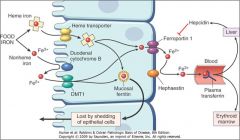
Important key players:
DMT1, Ferroportin, Transferrin, Ferritin, Hepcidin |
|
|
How many copies of α- and β-globin genes are there?
|
α-globin: 2 copies
β-globin: 1 copy |
|
|
What are the defective alleles associated with β-thalassemias?
|
β+ : splicing or promoter mutations leading to decreased β-globin synthesis
β0 : chain terminator mutations completely blocking β-globin synthesis |
|
|
What are the genotypes for β-thalassemia major?
|
β+/β+ or β+/β0 or β0/β0
|
|
|
What is the clinical outcome for patients with β-thalassemia major?
|
Severe tranfusion-dependent anemia.
|
|
|
What are the genotypes for β-thalassemia trait (minor)?
|
β+/β or β0/β
|
|
|
What are some major laboratory features of β-thalassemia major?
|
anisocytosis, poikilocytosis, microcytosis and hypochromia. Target cells, basophillic stippling, and fragmented RBCs are seen.
|
|
|
What is a major X-ray finding of patients with β-thalassemia major?
|
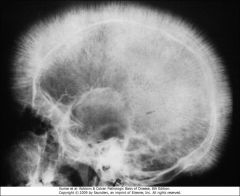
"Crew Cut" appearance due to extramedullary hematopoiesis
|
|
|
Are patients with β-thalassemia trait usually symptomatic?
|
No, but they may have mild anemia and some red cell abnormalities.
|
|
|
What is another word for β-thalassemia trait?
|
β-thalassemia minor.
|
|
|
Where are β-thalassemias most common?
|
Mediterranean countries, parts of Africa, and Southeast Asia.
|
|
|
What is the genotype for Hemoglobin H disease?
|
Deletion of three α-globin chains
|
|
|
What is hemoglobin H (HbH)?
|
A tetramer of β-globins which form due to low levels of α-globin.
|
|
|
Anemia in patients with a decreased TIBC, decreased serum iron, and decreased iron saturation?
|
Anemia of chronic disease
|
|
|
Anemia presenting with increased TIBC, decreased ferritin, decreased serum iron.
|
Mycrocytic, Hypochromic, usually secondary to iron deficiency. Also seen with Thalassemia, lead poisoning
|
|
|
Anemia in patients with an increased TIBC, decreased serum iron, and normal iron saturation?
|
Iron-deficiency anemia.
|
|
|
How is the diagnosis of immunohemolytic anemia made?
|
On the basis of a dirct Coombs antiglobulin test (patient's RBCs + anti-IgG antibodies → agglutination)
|
|
|
What are the obvious clinical signs of paroxysmal nocturnal hemoglobinuria, and how does this happen?
|
Dark urine in the morning due to increased complement activation during sleep when the CO2 level ↑ and thus pH ↓.
|
|
|
What are the two prinicple types of immune hemolytic anemia?
|
1. Warm autoantibodies (IgG)
2. Cold autoantibodies (IgE) |
|
|
What very important autoimmune disease may cause immune hemolytic anemia?
|
Systemic Lupus Erythematosus
|
|
|
What are the two major mechanisms of hemolysis?
|
Extravascular (immune mediated in spleen) and intravascular (mechanical) hemolysis.
|
|
|
What are some laboratory and clinical findings that distinguish between intravascular and extravascular hemolysis?
|
Intravascular: hemoglobinuria, dark urine, and urine hemosiderin.
|
|
|
What is disseminated intravascular coagulation (DIC)?
|
Activation of coagulation cascade and widespread deposition of fibrin throughout microcirculation. Often called consumption coagulopathy.
|
|
|
What is commonly seen in DIC?
|
Increased D-dimers and other fibrin split products.
|
|
|
What is thrombotic thrombocytopenic purpura (TTP)?
|
Defect in ADAMTS13 (cleaves vWF) leads to ↑ plasma vWF and widespread formation of platelet throbmi in microcirculation.
|
|
|
What is hemolytic uremic syndrome (HUS)?
|
E. coli 0157:H7 shiga-like toxin binds to endothelial cells in kidney and microvasculature, initiating platelet activation.
|
|
|
What major clinical findings differentiate TTP and HUS?
|
TTP: transient neurological defects
HUS: acute renal failure |
|
|
What does the prothrombin time (PT) measure?
|
Extrinsic and common coagulation pathways.
|
|
|
What does the partial thromboplastin time (PTT) measure?
|
Intrinsic and common coagulation pathways.
|
|
|
How can TTP and HUS be distinguished from DIC?
|
PT and PTT should be normal in TTP and HUS because coagulation cascade is not important in the mechanism.
|
|
|
What is immune thrombocytopenic purpura (ITP)?
|
Autoantibodies to gp IIb-IIa or Ib-IX obsonize platelets → phagocytosed in spleen.
|
|
|
What will you expect to see in the bone marrow in ITP?
|
Increase in the size and number of megakaryocytes in response to platelet destruction.
|
|
|
How is chronic ITP treated?
|
splenectomy.
|
|
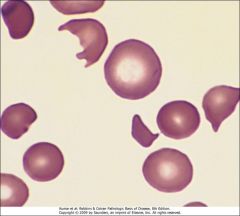
List several important disorders that are associated with schistocytes (helmet cells).
|
Mechanical heart valves, DIC, TTP, HUS, SLE.
|
|
|
What is reactive lymphadenitis?
|
Enlargement of the lymph nodes in response to acute or chronic infections.
|
|
|
What is infectious mononucleosis usually caused by?
|
Epstein Barr Virus (EBV)
|
|
|
What are two major pathological changes associated with infectious mononucleosis?
|
1. Reactive lymphadenitis
2. Atypical lymphocytes (Downey Cells) |
|
|
What is the most common inherited bleeding disorder?
|
Von Willebrand Disease
|
|
|
What is the major clinical feature of Von Willebrand Disease?
|
Prolonged bleeding time despite normal platelet count. May see ↑ PTT due to ↓ Factor VIII
|
|
|
What is the pathophysiology behind Hemophilia A and Hemophilia B?
|
Hemophilia A: Factor VIII deficiency
Hemophilia B: Factor IX deficiency |
|
|
Why are Hemophilia A and B clinically indistinguishable?
|
Because Factor VIII and Factor IX interact to activate Factor X. Thus deficiency in either VIII or IX will have the same outcome.
|
|
|
What are the clinical features of Hemophilia A and B?
|
Recurrent severe hemarthroses, easy bruising, massive hemorrhage after trauma, prolonged PTT.
|
|
|
How is Von Willebrand Disease differentiated from hemophilias?
|
Hemarthroses are uncommon in Von Willebrand disease.
|
|
|
What does bleeding time test?
|
The platelet response. Prolonged times indicate low platelet numbers and/or defective platelets.
|