![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
113 Cards in this Set
- Front
- Back
|
Differentials of Global Developmental Delay |
Chromosomal: T21, Prader Willi, Angelman's, Williams X-linked: Fragile X, Rett's AD: De novo, Noonans AR: Inborn errors of metabolism (Consanguinity) Mitochondrial: Multisystem (NARP - neuropathy, ataxia, retinitis pigmentosa, ptosis), (MERRF - myoclonic epilepsy w ragged red fibres) Prenatal: TORCH, Toxins, Radiation, Foetal alcohol syndrome Perinatal: Premature, Hypoxia, Infx, Trauma, IVH Postnatal: Trauma (Accidental/NAI) , Neglect, Malnutrition, Hypothyroidism, CNS haemorrhage, Hypoxia, Infx, Tumour |
|
|
Defn of Global Developmental Delay |
Significant Delay in 2 or more developmental domains: -Gross Motor -Fine Motor -Social -Speech and Language |
|
|
What Invx are done to diagnose Congenital Hypothyroidism? |
Screened for in the neonatal period Invx: Blood spot on a Guthrie Card - TSH^^, T4 ** - Radionucleotide scanning Tc99: to check if caused by dysgenesis or biosynthetic hormone disorder. |
|
|
How does Congenital Hypothyroidism present? |
Development: Growth plateaus GDD Basic fnc: Constipation Poor feeding Sounds: Hoarse cry Apperance: Prolonged Jaundice Protruding tongue Hypotonia (floppy baby) Umbilical hernia Coarse facial features Dry skin |
|
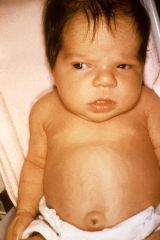
Spot Diagnosis Cause |
Congenital Hypothyroidism Cause: Athyreosis Hypoplastic thyroid gland Iodine deficiency |
|
|
Mgmt of Congenital Hypothyroidism Consequences if not treated |
Hormone replacement ASAP: Levothyroxine, inital dose 50mcg/kg/day Monitor: Keep T4 lvls in upper end of normal range. Keep TSH lvls in lower end of normal range Monitor serum TSH and T4 levels: Yr 1: every 1-2 mos Yr 2: every 2-3 mos >2 yrs old: every 4-6mos Consequences: Growth: Delayed, Short stature Motor: Ataxia, Hypotonia, Poor coordination Neuro: Mental ret. |
|
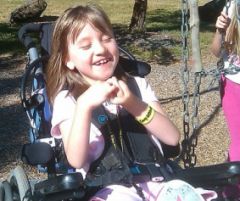
1) Spot Diagnosis 2) Cause |
Rett Syndrome- Note the wringing of hands, The high dependency wheelchair, Young age Cause: X-linked Mutation of MECP2 gene on Xq28 |
|
|
Rett Syndrome: Presentation |
Progressive disorder Girls Only Progress normal developmentally initially. When >1 yr old, massive neuro-developmental regression Loss of speech Loss of purposeful hand movements Stereotypic hand-wringing Erratic breathing: Breath holding + hyperventilation Scoliosis Seizures Prognosis: Death as a teenager |
|

Spot Dx Cause |
https://www.youtube.com/watch?v=U5J0kvFSTtA
Dx: Angelman syndrome - Note smiling, widely spaced teeth, uplifted arm, strabismus Cause: Impaired/Absent fnc of maternally imprinted UBE3A gene on Chr 15 |
|
|
Angelman Syndrome: Presentation |
GDD Type of Autism Behaviour: Always smiling, Inappropriately happy, Excitable, hand flapping Speech: No functional speech Motor: Wide-based ataxic gait, Hands upheld when walking, Appearance: Disproportionately small head (maybe microcephaly by age 2) Seizures by <3yrs |
|
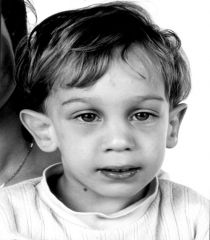
Spot Dx Cause |
Fragile X Syndrome Cause: full expansion (>200 repeats) of (CGG)n triplet repeat in FRAXA gene on chr Xq27 XD |
|
|
Fragile X: Presentation |
https://www.youtube.com/watch?v=mWjfYOmxjO0 GDD Males (some females) Most commonly inherited cause of mental impairment and autism GDD Behaviour: Hand flapping, Gaze avoidance, Resistance to changes in routine, Social anxiety Appearance: Long face, Large floppy ears, Hypotonia Other physical: Hyperextensible fingers, High arched palate, Postpubescent macro-orchidism Learning: Anything from normal IQ --> Severe intellectual disability
|
|
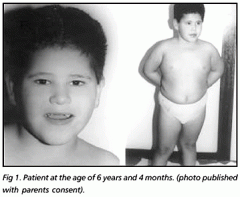
Spot Dx Cause |
Dx: Prader Willi Syndrome Cause: Disruption to paternally derived imprinting of chr15q 11-13 |
|
|
Prader Willi: Presentation, How is it diagnosed |
Floppy Feeding Difficulties Growth: Initial FTT --> Rapid weight gain from 1-6yrs Appearance: Short stature, Truncal obesity Behaviour: Insatiable appetite, Food foraging GDD + learning disability Diagnosed: Molecular genetic analysis |
|
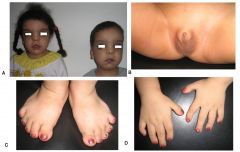
Spot Dx Cause |
Dx: Bardet Biedl Cause: AR, 8 different genes identified |
|
|
Bardet Biedl: Presentation |
Physical: Obesity, Polydactyly/Syndactyly/Brachydactyly, Hypogonadism (boys)/Genitourinary malformations (girls) Kidneys: Renal failure Eyes: Retinitis Pigmentosa |
|
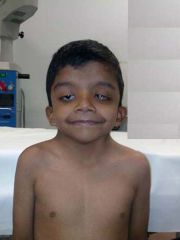
Spot dx Cause |
Dx: Noonan Syndrome (looks like turners in boys Cause: AD, but 80% are sporadic Mut genes affecting RAS-MAPK path Genetically heterogenous Around 50%= Mutation PTPN11 gene on chr 12q |
|
|
Genetic Disorders causing Obesity |
Prader Willi Syndrome Bardet-Biedl |
|
|
Genetic Causes of Short Stature |
Turners syndrome Noonans syndrome Congenital Hypothyroidism |
|
|
Genetic Causes of Tall Stature |
Marfan's |
|
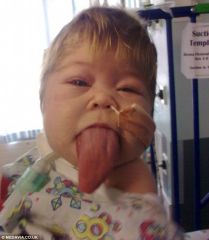
Spot Dx Cause |
Dx: Beckwith Wiedemann Syndrome Cause: Disruption of imprinted region on chr11p15 |
|
|
Beckwith Wiedemann: Presentation |
https://www.youtube.com/watch?v=NA16opHKpHQ As they grow: Normal size and intelligence achieved At birth: Polyhydramnios, Macrosomia, PPROM Port wine stain Macroglossia Neonatal hypoglycaemia Midline abdominal wall defects (umbilical hernia / omphalocoele/ exomphalos Ear creases/pits Cancer: Hepatoblastoma, Wilm's tumour, Gonadoblastoma |
|
|
Noonan Syndrome: Presentation |
https://www.youtube.com/watch?v=nlvSqVgIrdk GDD Gross appearance: Webbed neck, HYpertelorism (widely spaced eyes), ptosis, proptosis, low set backward rotated ears, triangular face, deeply grooved philtrum Cardiac: Pulmonary valvular stenosis, ASD, HCM GU: Crypt-orchidism GI: FTT, Swallowing difficulties, Gastroparesis Lymphatic: Posterior cervical hygroma (web), Lymphoedema Bleeding disorders Hearing loss Development: GDD, Intellectual disability, Autism, Hypotonia, Difficulty articulating |
|

Spot Dx Cause |
Dx: Williams Syndrome Cause: Microdeletion on chr 7q11 that involves elastin gene |
|
|
Williams Syndrome: Presentation How do you diagnose it? |
GDD Electrolyte: HypERcalcaemia (15%) Behaviour: Overly friendly, Short attention span, Anxiety Cardiac: Supravalvular aortic stenosis, Peripheral pulmonary branch stenosis Appearance: Periorbital fullness, Full cheeks, Wide mouth, Small widely-spaced teeth Diagnosis: FISH for chr7q11 microdeletion |
|
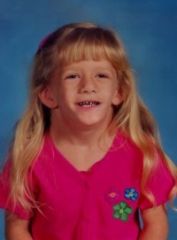
Spot dx Cause |
Dx: Turner Syndrome Cause: 45XO Mosaicism 45XO/ 46XX |
|
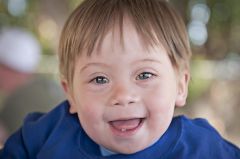
Spot dx Cause |
Dx: Down Syndrome Cause: Trisomy 2, mostly due to non-disjunction during maternal oogeneisis |
|
|
Turner syndrome: Presentation |
Because majority have mosaicism, many won't present with stereotypical features. Girls only Short stature: Growth rate decreases between 3-5 years, don't get a pubertal growth spurt Ovarian dysgenesis, gonadal failure, amenorrhoea Appearance: Low hair line, Low set ears, Shield chest, Lymphoedema, Cystic hygroma --> webbed neck, Shortened metacarpal 4 Cardiac: AV stenosis, Coarctation of aorta, Bicuspid aortic valve Behaviour: ADHD sometimes |
|
|
Turner syndrome: Mgmt |
SC daily injections: rhGH 10mg/m2/wk Oestrogen to induce puberty Alternative: Combo therapy: Oe + oxandralone (anabolic steroid) to improve height. |
|
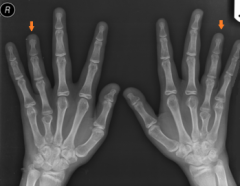
Spot diagnosis |
Dx: Turners syndrome Shortened IV metacarpal |
|
|
Down Syndrome: Presentation |
GDD Tone: Generalised hypotonia, Marked head lag Face: Prominent epicanthic folds, Brushfield spots, Small low set ears, Protruding tongue, Flat facial profile. Short neck Digits: 'Sandal gap' between 1st and 2nd toes, wide hands, short fingers, single palmar crease Assoc conditions: Cataracts, Deafness, DDH, Eczema, Cardiac: 40-50% congenital heart disease (AVSD, ASD, BSD, TOF) GI: Duodenal atresia, Hirschsprungs Leukaemia Acquired hypothyroidism |
|

Spot Dx |
Dx: Down syndrome - wide sandal gap between 1st and 2nd toes. |
|
|
Down Syndrome: Mgmt |
Cardiac: Refer for detailed cardiac assessment Deafness: Refer to audiology DDH: Refer to orthopaedics for hip US LT monitoring by MDT Physio: TO improve posture and tone Hypothyroid: TFTs annually. Audiology + ophthalmic assessmentevery 1-2yr Genetic counselling Put parents in contact with support organisation |
|
|
Down Syndrome: Prognosis |
Death by congenital cardiac defect Alzheimers by 40yrs |
|
|
1st line invx for Global Developmental Delay? |
Karyotype- Fragile X, Turner, FBC, Feritin, Iron studies - Malnutrition, Infx RFTs - Bardet Biedl Calcium - Turners =hypercalcaemia TFTs - Congenital hypothyroidism CK - Duchenne Muscular Dystrophy Vision and hearing assessment: Down, Noonan |
|
|
2nd line invx for Global Developmental Delay? |
Neuroimaging - Tuberous sclerosis etc EEG - Rett syndrome Metabolic - Hypothyroid, IEOM Infx screen - TORCH Further genetic studies - FISH for Williams, Molecular genetic studies for Prader Willi. |
|
|
Age correction for prematurity: For how long? |
Until the child is 2 years old. |
|
|
GDD: MGMT? |
Full history and exam - Regression? Every attempt should be made to identify an underlying cause Correct for prematurity Assess the extent of the delay in each area Invx Refer to a child development team assessment Treat the underlying cause Early Intervention Services/MDT: PT, SALT, OT IFSP - Individualised Family Structure Program |
|
|
Causes: Speech and Language Delay |
-Maturational Language Delay -Developmental Language delay -Specific language impairment Articulation disorder: Clef lip, Cleft palate, Fluency disorder: Stammer Communication difficulty: ASD -GDD -Hearing loss -Prematurity / LBW Infx: Intrauterine, HIV, Meningitis Neurological: CP, Epilepsy Toxic: Lead Metabolic: PKU, Hypothyroid Genetic: T21, Fragile X, Williams, Angelman, TS, NF1 FHx Environment: Deprivation, Neglect -Selective mutism -Verbal apraxia |
|
|
Definition of Low Birth Weight? |
Liveborn infant with birth weight of <2.5kg |
|
|
Rett Syndrome: Characteristic EEG |
1) Background slowing 2) Central, short duration spikes 3) ^^epileptiform activity during sleep |
|
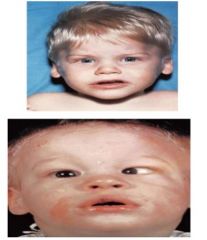
Spot Dx Cause |
Dx: PKU Cause: IEOM, due to lack of the enzyme Phenylalanine Hydroxylase --> can't break down phenylalanine --> accumulation |
|
|
PKU: Presentation |
Musty odour to sweat and urine Hypopigmentation: Pale skin, Blonde hair Eczema Later: Seizures, Developmental Delay Hyperactivity Learning disability |
|
|
PKU Mgmt |
Diet: Eliminate foods high in phenylalanine (lobster, soybeans, chicken, fish spirulina, nuts) Keep a food diary Limit starchy foods (potatoes, corn) Med: Tetrahydrobiopterin (cofactor B4) |
|
|
IEOMs producing strange smells: |
1) MSUD: Urine smells of sweet maple syrup 2) PKU: Sweat and urine = musty odour 3) Tyrosinaemia 1: Rotten fish/cabbage |
|
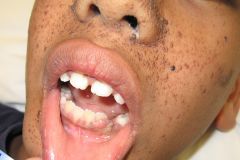
Spot Dx Cause |
Dx: Tuberous sclerosis Cause: AD, but 2/3rds are sporadic TSC1 and TSC2, both tumour suppressor genes, are mutated |
|
|
Tuberous Sclerosis: Diagnostic criteria |
DX = (2 major OR 1 major + 2 minor criteria) Major Skin: Shagreen patch Hypomelanotic macules (>3) Brain: Subependymal giant cell astrocytoma Subependymal nodules Head: Facial angiofibromas Retinal nodular haematoma Other: Ungual fibroma Cardiac rhabdomyoma Minor: Skin: Confetti skin lesions Bone: Bone cysts Brain: Cerebral WM migration tracts Head: Gingival fibromas Pits in dental enamel Retinal achromic patch Renal: Multiple renal cysts Other: Non-renal haematoma Rectal polyps |
|
|
Tuberous Sclerosis: MGMT |
Symptomatic, depending on the organ systems affected Expert Assessment: Recurrence risk in family Expert Assessment: Seizures (especially with West syndrome) Cardiology: Rhabdomyoma Renal: >9yrs = biannual renal US with regular enquiry for loin pain/renal function Resp symps: Screen for pulmonary lymphangiomas (only in girls) Ophthalmology: Ophthalmologic haematomas |
|
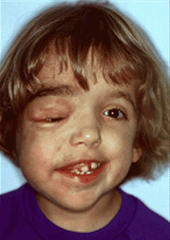
Spot Dx Cause |
Dx: NF1/ Peripheral NF/Von Recklinghausens Cause: AD Mutation of NF1 gene on chr17 |
|
|
NF1: Diagnostic criteria
|
At least 2 of the following: >6 café au lait macules >5mm diameter before puberty OR >15mm diameter after puberty Skin fold/Axillary freckling Skeletal dysplasia 1 Neurofibroma/plexiform neurofibroma 1 Lisch nodule in iris Optic glioma Affected 1st degree relative |
|
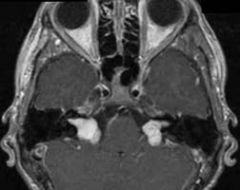
Spot dx Cause |
Dx: Bilateral vestibular schwanomas- associated with NF2 Cause: NF2 mutation on chr22 |
|
|
NF2: Diagnostic Criteria |
1 major OR 2 minor of the following: Major -Bilateral acoustic schwannomas -Unilateral acoustic schwanoma + 1st degree relative with NF2 Minor: MEGS Cat Meningioma Ependymoma Glioma Schwannoma Cataracts |
|
|
List AD disorders |
Achondroplasia Ehlers Danlos Familial hypercholesterolaemia Huntingtons Marfans Myotonic dystrophy Neurofibromatosis Noonan's Osteogenesis Imperfecta Otosclerosis Polyposis coli Tuberous Sclerosis |
|
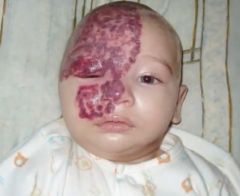
Spot dx Cause |
Sturge Weber syndrome Cause: Sporadic |
|
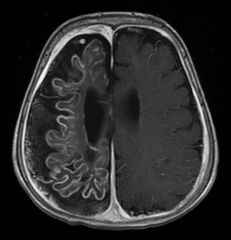
Spot Dx: Cause: |
Dx: Leptomeningeal angiomatosis Cause: Sturge Weber syndrome (sporadic) |
|
|
Sturge Weber Syndrome: Presentation Mgmt for associated epilepsy |
Port wine stain in distribution of ophthalmic branch of trigeminal nerve Leptomeningeal Angiomatosis intracranial lesion Severe: Intractable epilepsy, hemiplegia, learning disability Less severe: Deterioration rare past 5 yrs of age. Seizures and learning difficulties may still be present. Mgmt: Intractable epilepsy = Hemispherectomy |
|

Spot Dx Cause Epi |
Duchenne Muscular Dystrophy
Cause: XR Xp21 mutation of gene dystrophin Absence of dystrophin --> progressive muscle cell damage Epi: Commonest disabling neuromuscular disorder of childhood. 1/4000 males |
|
|
Commonest disabling neuromuscular disorder of childhood is....? |
Duchenne Muscular Dystrophy |
|
|
DMD: Presentation |
Diagnosed at 4-5yrs Gross motor delay Clumsiness Waddling gait Proximal myopathy Gower's sign Calf pseudohypertrophy Reduced/absent reflexes |
|
|
DMD: Invx: Prognosis |
Invx: ^^ CPK: Creatinine phosphokinase Genetic studies: Xp21 dystrophin mutation EMG (elctromyography): Shows myopathic destruction of tissue Muscle biopsy: absent dystrophin on immunochemistry Prognosis Walking frame aged 8-10. Wheelchair aged 10-14 Cardiomyopathy + Resp problems Scoliosis Cognitive decline Death = early 20's from heart/lungs |
|
|
Mgmt of NMJ and Muscular Disorders |
Assessment of power, jt ranges, contrractures PT and OT advice SALT and dietician access Manage resp/cardio snd other system compx Liaison with education/social services Genetic counselling Psychological support |
|
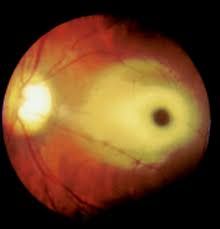
Dx Cause |
Dx: Cherry red spot on the macula Causes: Tay-Sachs Niemann-Pick |
|
|
Tay Sachs: Cause Epi |
Enzyme defect: Hexosaminidase A AR Epi: Ashkenazi Jews |
|
|
Niemann Picks: Cause
|
Enzyme defect: Sphingomyelinase |
|
|
Gaucher's Disease: Cause Epi |
Enzyme defect: Beta glucosidase Epi: 1/500 Ashkenazi Jews |
|
|
Tay Sachs: Presentation Prognosis Dx |
Regression: Developmental regression in late infancy Startle: Exaggerated startle response Vision: visual inattention cherry spot in macula Social: Unresponsiveness Hypotonia severe Appearance: Enlarging head Prognosis:Death by 2-5yrs Dx: Measuring specific enzyme activity of Hexosaminidase A Prenatal diagnosis and Carrier detection of high risk couples |
|
|
Niemann Picks: Presentation Mgmt Dx
|
Presentation: Chronic childhood form: Splenomegaly Bone marrow suppression Normal IQ Acute infantile form: Splenomegaly Neurological degeneration Seizures Mgmt: Enzyme replacement Hypersplenism: Splenectomy Dx: Prenatal detection and Carrier detection of high-risk couples |
|
|
Niemann-Pick Disease: Presentation Prognosis |
Presentation: Hepatosplenomegaly: **appetite, abdo distention, pain, FTT Splenomegaly: Thrombocytopenia CNS accum: Ataxia, Dysarthria, Dysphagia BG accum: Dystonia( Abnormal posturing) Cerebral accum: Seizures, Dementia Sleep disorders Weak bones Cherry red spot in macula Deteriorating vision and hearing Prognosis: Death by 4yrs |
|
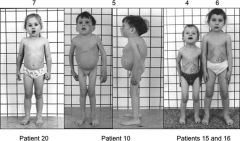
Spot dx Cause |
Dx: Gaucher's disease (note the spleomegaly, bone involvement with ostteoporotic-looking back posture) Cause: Enzyme defect- beta-glucosidase |
|

Spot Dx (of the back twin) Cause: |
Dx: Niemann Pick disease (note the hepatosplenomegaly protruding, FTT in comparison to sister, hypotonia( tongue out), lid lagging (supranuclear gaze palsy) Cause: Sphingomyelinase |
|
|
DMD vs BeckerMD- Compare |
1 = Duchenne 2=Becker Inheritance 1) XL 2) XL Dystrophin: 1) Completely non-functional protein 2) Partially functional protein / reduced amt Diagnosis method: 1) Whole blood DNA / EMG / CPK^^/ Muscle biopsy 2) Immunostaining Clinically evident: 1)3-5 yrs 2) >10yrs Cardiac involvement: 1) DCM and/or resp failure 2)Rare Ambulatory: 1) until 9-12 2) until 18yrs+ Life expectancy: 1)16-19 yrs 2) Twice as long Course: 1) Quick, regular, progressive proximal weakness 2)Milder, slower course Other fx: 1) Calf pseudohypertrophy, Worsening contractures + scoliosis 2)Calf pseudohypertrophy + Pes cavus |
|
|
Is corticosteroid therapy effective Rx for DMD? |
Yes- improved strength with optimal dose of Prednisone 0.75mg.kg/day Strengthening effects last for 3 yrs BUT side effects (weight gain, ^^ susceptibility to infx) may outweigh benefits in many cases |
|
|
Most likely diagnosis: Child with progressive walking difficulties evolving over several days? |
Guillain Barré Syndrome |
|
|
How is Down Syndrome diagnosed prenatally? |
1) Detailed US scanning 2) Amniocentesis (wk 15+) (1% risk miscarriage) 3) Chorionic villus sampling (wk 11-13) (>1% risk miscarriage) 4) Foetal blood sampling Positive for Down: Chromosomal analysis Nuachal thickening (fat pad) Dudodenal atresia AV canal defect of heart |
|
|
When would you suspect IEOM? How do IEOMs present? |
Suspect: Positive family hx FHx of unexplained deaths Present: General: Severely sick w/o adequate explanation Feed: FTT, Poor feeding, Persistent vomiting Liver: Jaundice, Hepatomegaly Neuro: Lethargy, Coma, Convulsions Scent: Unusual odour of body/urine Appearance: Dysmorphism |
|
|
How does Galactosemia present? How to treat Galactosemia? Cause of Galactosemia Consequence of not recofnised and treated promptly |
Feeding: Vomiting, Diarrhoea, FTT Liver: Hepatomegaly and liver dysfunction Blood: Hypoglycaemia Eyes: Cataracts in oil-drop pattern Kidneys: RTA Treat: Galactose free diet (soy substitution) Cause: Deficiency of G1PUT (galactose 1 phosphate uridyltransferase) Consequences: Death by E.Coli infx, Ovarian failure, Mental ret, |
|
|
Presentation of MCADD? Mgmt of MCADD |
Present: Nonketotic hypoglycaemia Myopathy Cardiomyopathy Hyperammonemia MGMT: Frequent feedings of high carb, low fat diet Carnitine supplementation during acute episodes. |
|
|
Give an EG of the following types of IEOMs a) Defects in AA metabolism b) Defect in Carb metabolism c) Defect in Fatty Acid Oxidation d) Defect in Metal metabolism e) Defect in heme pigment biosynthesis f) Lysosomal storage disorders - Mucopolysaccharidoses g) Lysosomal storage disorders- non-muco |
a) PKU, MSUD b) Galactosemia c) MCADD d) Wilson's disease e) Porphyrias f) Hurlers, Hunters g) Niemann picks, Tay Sacchs, Gauchers, Metachromic leukodystrophy |
|

Spot Dx Inheritance |
Dx: Hunter's Syndrome Inheritance: XR |
|
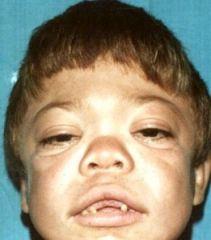
Spot Dx Inheritance |
Dx: Hurlers Syndrome Inheritance: AR |
|
|
Hurlers vs Hunters: Distinguish Common features |
A = Hurler B= Hunter Hurler = more severe Begin to present with symptoms A= After 1 yr B: 2-4yrs old Papules A= No B= Yes, over scapula, shoulder, lower back Corneal clouding A= Yes B = No Prognosis A= Death 10-15 yrs B= Death by 20 Mgmt: A = early bone marrow transplant to prevent neurodegeneration B = None Common: Hepatomegaly, Progressively stiffer and more contracted joints, |
|
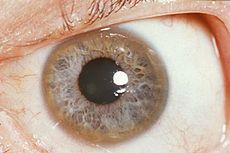
Spot Dx Cause Diagnosis- How? Present Mgmt |
Dx: Kayser FLeischer ringsm seen in Wilsons disease Cause: AR defect in copper excretion. Copper deposits in liver first, followed by brain, eyes, heart Diagnosis: ** serum ceruloplasmin. ^^urine copper Present: Liver: Hepatic dysfnc Eye: Kayser Fleischer Rings Brain: Behavioural change, Seizures, Dysarthria, Dystonia, Ataxia, Tremors, Mgmt: 1) Avoid copper containing food (nuts, choc, shellfish) 2) Chelation therapy with oral penicillamine and Zn salts. 3) Liver transplant |
|

Spot Dx Cause Diagnosis- how |
Dx: Menkes kinky hair syndrome Cause: XR, abnormal copper transport ==> Low copper lvls Present -Myoclonic seizures -Pale, kinky, friable hairoptic nerve atrophy -severe mental ret -progressive neuro degen Diagnosis: Low serum ceruloplasmin Low serum copper Characteristic hair |
|
|
Outline basic language milestones from birth to 1 yr old |
Birth: Attune to human voice Develops differential recognition of parents voices 2-3mos: Cooing Musical sounds (ooh ooh, aah) 6mos: Babbling (mixing vowels and consonants) 9-12mos : Jargoning (inflection , cadence) Mamma Dadda nonspecific 12 mos: 1-3 words mamma dadda SPECIFIC |
|
|
Outline basic language milestones from 18 mos to 3 yrs |
18mos: 20-50 words 2 phrase sentence 2 yrs: 2 word telegraphic sentences (mommy come) 25-50% of child's speech intelligible 3yrs: 3 word sentences >75% should be intelligible |
|
|
What parts of a Hx would you focus on: child with speech delay |
1) Parental concerns 2) Parental education 3)RFs for hearing loss : perinatal exposure to infections, compx (prolonged jaundice, TORCH, <1500g, aminoglycosides) 4) Developmental hx 5) Fhx of language delay / hearing loss 6) How many languages are spoken at home? |
|
|
Examine: Child with speech delay |
Centiles: Kleinefelter? FTT? Appearance: Dysmorphic? Autism: Social interaction Ear: Tempanic membrane normal? intact? Mouth: Check that they can produce speech. Palate? Teeth? Drooling? Bifid uvula Neuro exam Skin: Shagreen patches / Ashleaf / Neurofibromas, Bruising |
|
|
DDX for speech delay |
GDD Genetic: Down, Fragile X, William,TS CP Conditions Epilepsy Hypothyroid PKU
TORCH, HIV, Meningitis Jaundice Aminoglycosides Noonan, Down, Niemann Pick Environmental deprivation/exposure Lead Pervasive developmental disorders : Autism, Other: Selective mutism Verbal |
|
|
https://www.youtube.com/watch?v=szjfC9K190U
Watch and diagnose |
Verbal apraxia |
|
|
Outline pathophysiology of stuttering |
1) Child experiences blocks and repetitions (learned mechanisms) 2) Fear of repetition ==> unease 3) Anxiety and high allostatic stress load 4)Anxiety ^^ stuttering Normal speech: When relaxed and speaking spontaneously without planning |
|
|
Causes of Stuttering |
Acquired = Rare Head trauma/Tumur / Stroke / Drugs Genetic Anatomical abnormalities Dopamine abnormalities Developmental: Occurs at 3yrs when child's thought processes outstrip ability to express themselves. Often improves if not too much attention is paid to it. |
|
|
Mgmt of Stuttering |
1) Diaphragmatic breathing 2) Operant conditioning: Fluency shaping therapy 3)Stuttering modification therapy 4) Fluency electronic device 5)Pagoclone: Meds 6)Speech therapy 7) Support groups |
|
|
Prognosis: Stuttering |
75% recover by teenage years. Girls > boys recover |
|
|
Hearing Loss: RFs |
FHx Drugs: Ototoxic - aminoglycosides Prolonged jaundice Premmie, LBW Infx: TORCH (especially CMV), HIV, Mumps HIE Craniofacial abnormalities |
|
|
Hearing Loss: Causes |
Genetic: Non-syndromic 80% Connexin 30 and 26: DFNA (AD), DFNB (AD, DFNX (XL) Genetic: Syndromic 20% AD:Stickler Waardenburg: SN deafness + pigmentation anomalies Treacher Collins: Conductive deafness + midface hypoplasia AR: Pendred: SN deafness + hypothyroid Usher: SN deafness + Retinitis pigmentosa XL: Alport: SN deafness + renal failure (+ pyelonephritis + haematuria) Mitochondrial Infx: CMV, Rubella Cochlear aplasia |
|

Spot Dx Main fx |
Waardenburg syndrome Fx: SN deafness Pigmentation anomalies |
|

Spot Dx Cause main Fx |
Treacher Collins Cause: AD. Commonly a TCOF1 gene mutation (and mutation of other genes..) ==> ** production of rRNA ==> abnormal facial bone formation Main Fx: -Midface hypoplasia (no cheek bones) -Conductive hearing loss -Cleft palate -Mandibular hypoplasia ==> airway problems |
|
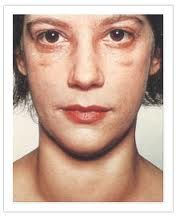
This woman also has SN hearing loss. Spot dx? |
Pendred syndrome: SN hearing loss + hypothyroid |
|
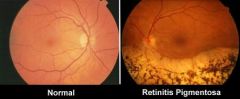
This patient has RP and hearing loss Spot dx? |
Dx: Usher syndrome Retinitis pigmentosa + SN hearing loss |
|
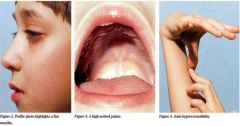
Spot Dx Main Fx |
Stickler syndrome Fx: Collagen disorder SN hearing loss Ocular problems High arched palate Joint issues |
|
|
DDx: Visual + Hearing problems in a child |
1) Usher syndrome 2) Stickler syndrome 3) Down syndrome 4) CHARGE syndrome 5) Juvenile Alport syndrome 6) CMV infx 7) Rubella infx |
|
|
1ary invx for hearing loss |
1) Hx: HxPC, FHx 2) Exam: Clinical and Developmental 3) Audiology: all of the family 4) ECG IF FHx of sudden death 5) Ophthalmology (Usher, Stickler, CHARGE..) 6) Urinalysis: Haematuria (alports) 7) Connexin 26,30 8) MRI brain |
|
|
Defn of delayed walking? Average age that children begin to walk? When should you refer for further opinion? |
Defn: Not walking by 18 mos Average: 13 mos Refer: 18 mos if not walking |
|
|
Ddx delayed walking: |
Delayed motor maturation (familial) GDD (dysmorphic / microcephaly) Lack of opportunity (illness/ kept in a cot) Hypertonia: Cerebral palsy Muscular dystrophy: Duchenne's/ Becker MD |
|
|
Delayed Walking: Invx? |
If isolated delayed walking: CPK level only / Genetic studies / EMG / Muscle biopsy If evidence of cerebral palsy / hypotonia? CP: MRI of pyramidal tracts and BGs |
|
|
Ddx: Hypotonia |
Chr: Down Syndrome, Prader Willi, Noonan Lysosome storage: Tay Sacchs, Niemann Pick XL: Fragile X Metabolic: Congenital hypothyroidism CTD: Marfan, Ehlers Danlos Charcot Marie Tooth Cerebral palsy |
|
|
Short stature: Ddx |
Familial: MPH. SMall parents
- Commonly familial - Assoc with dieting/exercise -Normal puberty, but late - Delayed bone age IUGR/Premmie: 1/3 stay small Psychosocial: Catch up with nurturing Endocrine Cushing Hypothyroid GH: Deficiency/Resistance (pan-hypopituitarism) - Congenital Midline defects - Craniopharyngioma/Pituitary tumour - Meningitis - Cranial irradiation Chronic disease: CF, Congenital heart disease, Coeliac, IBD, CKD Genetic: Turners Noonans T21 Russell Silver SKeletal dysplasia (NB SITTING height) Achondroplasia (FGFR3 mutation) Scoliosis Metabolic storage disorders |
|
|
Tall stature: Ddx |
Familial (MC) Obesity - Advanced puberty but early closure of epiphyses Excess hormone Thyroid, Sex steroid, Adrenal steroid GH Genetic Long legged: Marfan, Homocystinuria, Kleinefelter Proportional: Beckwith Wiedemann, Sotos |
|
|
Short Stature: Invx |
• Bone Age (XR not Bone Scan) – Constitutional, Endocrine
• FBC – Anaemia • Renal – CKD • CRP – Chronic Disease • TFTS – Hypothyroidism • Anti-tTG – Coeliac • Karyotype – Turner (Female) • GH Provocation Testing (Insulin Tolerance Test) – GHDeficiency • MRI Brain if Neuro S&S – Tumour • FOLLOW UP MEASUREMENTS |
|
|
How to calculate mid-parental height? |
Girl: [Mum + (Dad - 13cm) ] /2 +/-8.5cm = range Boy [(Mum + 13) + Dad] /2 +/-8.5cm = range |
|
|
Defn: Short stature Tall stature |
Short: <3rd centile Tall: >98th centile |
|
|
Which hormones, in excess, can cause tall stature |
Sex steroids Thyroid hormone Adrenal steroid GH |
|
|
Tall stature: Exam and Invx |
Arm span - Marfan's Sub-ischial leg length- Marf, Homocystin, Kleine Mid parental height Serial height measurements- chart growth velocity BMI: Obesity Bone age: May be advanced (obesity) Karyotype: Chromosomal, XXY, XYY Genetic testing: Klinefelter, Marfans Homocysteine lvls: If mental ret (homocystinuria) Marfans suspected: Echo + assess aortic root Basal gonadotropins (LH,FSH,hCG - pubertal abnormalities) Sex hormone levels |