Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
61 Cards in this Set
- Front
- Back
|
What is the role of the nucleolus in the biogenesis of the ribosomal machinery?
|

NCL1 and NPM1 bind pre-rRNA
|
|

Analyze this data.
|
-Triton causes membrane to vesiculate
>causes transmembrane proteins to be released into supernatant (integral components) -NaOH causes peripheral proteins to be released into supernatant -huge detergeant resistant structure of RBC membrane |
|
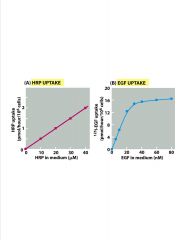
What are these 2 graphs showing?
|
-2 patterns of protein uptake in cells
>A=non-specific >B=receptor-mediatedlevel-off due to receptor saturation |
|
|
Describe the endocytosis of EGFr.
|
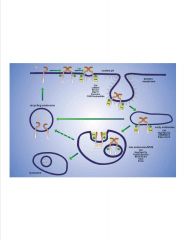
-EGF= Y kinase (autophosphorylated)
|
|
|
Describe how the clathrin coat is recruited.
|
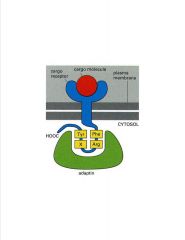
-intracellular C-terminal tail has signaling peptide recognized by apdaptin (brings in clathrin coat)
|
|
|
What is responsible for pinching off the clathrin-coated vesicle?
|
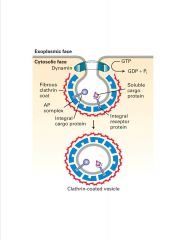
-dynamin=G protein; allows budding of vesicles
-gold particled dynamin EM micrograph |
|
|
How is a receptor targeted for clathrin-mediated endocytosis?
|
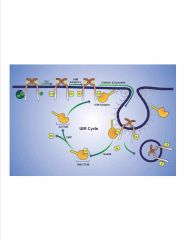
-ubiquitin recognized by UIM-adaptor
-ubi=8 kD protein added to substrate to target for degradation (usually) >E1, 2, and 3 are enzymes needed to ubiquitinate >Cbl puts ubi on >Nedd4=HECT E3 >UBP=de-ubiquitinater >UIM addapter removed by its own ubi (these proteins are part of adaptin complex |
|
|
What are 3 fates of bound ligand?
|
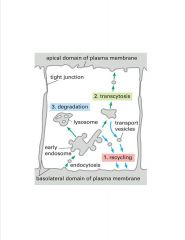
-polarized epi cell
>2 PM domains >recycling, degredation, and transcytosis are 3 options for a bound ligand |
|

Interpret this data.
(transferrin on left, EGFr on right) |
-fluorescently labeled protein added to cell to visualize localization
>transferrin disappears more quickly than EGF >this is b/c transferrin is recycled (after Fe release) whereas EGF is degraded |
|
|
Describe the uptake of iron into a cell.
|
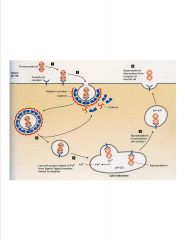
-transferrin maintains Fe conc. In cells
3) uncoating clathrin and adapters recycled back to surface 4) Late endosomes are acidic >apotransferrin binds receptor under acidic conditions whereas transferrin only binds at neutral pH 5) Apotransferrin released at neutral pH **neither transferrin nor receptor are degraded** -transporter in endosome shuttles Fe to cytosol |
|
|
What dictates the destination of vesicles?
|
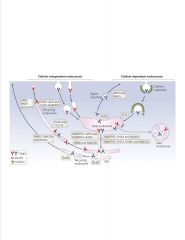
-Rab proteins keep endosomes going on a specific path
-each Rab has an effector >disctates fusion and docking of vesicles |
|
|
What components tether a vesicle to a membrane?
|
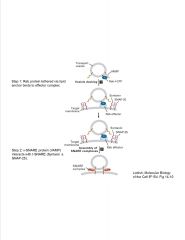
|
|
|
What components are involved in membrane fusion?
|
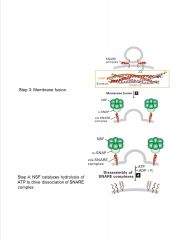
-late and early endosomes tend to fuse
-late endosomes and lysosomes tend to fuse |
|
|
What are 3 fates of endocytosed receptors?
|
Proteins that are internalized by receptor-mediated endocytosis may be either transcytosed, recycled, or degraded with or without their receptors.
Those ligands that are recycled (transferrin) transit only the early endosome while ligands (EGF and LDL) destined for lysosomal degradation transit both early and late endosomes. |
|
|
What components are required for vesicular trafficking?
|
Vesicular trafficking requires GTP, cytosolic GTP-binding proteins (RAB’s), cytosolic SNAP’s, NSF and membrane-bound SNARE’s.
|
|
|
Describe the late endosome.
|
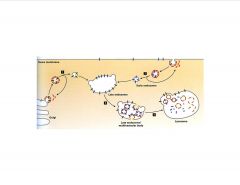
-late endosome tends to target for degradation
-recycling usually occurs in early endosome -Y kinase of EGFr is cytoplasmic in late endosome >in multivesicular body, kinase aspect is internalized |
|
|
What factors are needed for the invagination of the late endosome for receptor degradation?
|
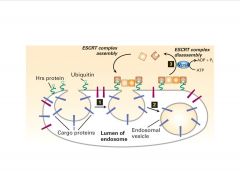
-ESCRT complex-mediated budding of multi-vesicular body
>doesn’t utilize clathrin or calveolin >Ubi stil required (mono) |
|
|
How does endocytosis play a role in thyroid metabolism?
|
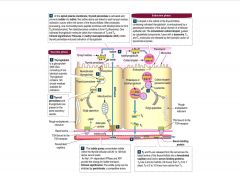
-thyroxine synthesized as thyroglobulin
>Y moieties of thyroglobulin cross-linked at apical surface, then stored in colloid >Y iodonated also >cell endocytoses colloid and degrades everything but T3 and T4 |
|
|
How does endocytosis play a role in cholesterol metabolism?
|
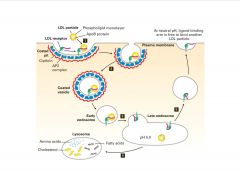
-LDL disocciates from receptor in late endosome due to acidity
>cholesterol released for membrane integrity |
|
|
What is the basis of familial hypercholesterolemia?
|
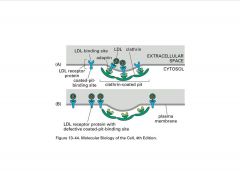
-hypercholesterolemia (familial) all based on receptor problems
B) 4 aa seq missing from LDLr >no apater recognition=no endocytosis |
|
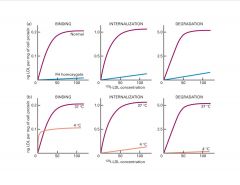
Interpret this data.
|
b) At 4C, just see binding b/c there’s no internalization or degradation at this temp (shows receptor #)
>at 15C, internalization doesn’t reach lysosomes |
|
|
Contrast how endocytosis fx the different components covered in this lecture.
|
Endocytosis down regulates tyrosine kinase receptors, up regulates thyroxine production, and modulates the intracellular transport of needed iron and cholesterol.
|
|
|
What are the lysosomal functions?
|
Degradation of endogenous and exogenous macromolecules and recycling of monomeric subunits (e.g., amino acids)
Degradation of aged or damaged organelles Removal of intracellular pathogens Homeostasis of iron and cholesterol Antigen presentation (e.g., macrophages) Bone remodeling (e.g., osteoclasts) Cell survival and Cell death |
|
|
How is lysosomal pH determined?
|
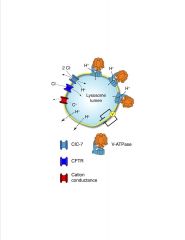
Determinants of lysosomal pH. Lysosomal acidification is dependent on V-ATPase, a large multimeric enzyme complex that transforms the energy of ATP hydrolysis into the movement of protons across the lysosome membrane. Electrogenic proton transport creates an electrical gradient that must be dissipated to establish the substantial chemical proton gradient. Electroneutrality can be maintained through the parallel influx of anions alongside protons. ClC-7, a chloride proton antiporter, and CFTR have been proposed to constitute the counter ion pathways in the lysosome membrane, as described in the text. The efflux of cations (C+) through distinct channels or transporters can also occur. Parallel proton leak pathways (dotted lines) are also known to exist and require continued V-ATPase activity to maintain a steady-state pH. Acidification kinetics are also contingent on the luminal buffering power (not depicted).
-V-ATPase is similar to F1/F0 ATPase of mitochondria >pump requires ATP >other ion channels modulate pH -many membrane proteins of lysosomes have COOH moieties that become salicylic acids that prevent the protein from being degraded by hydrolytic enzymes |
|
|
Compare/contrast protein vs. carbohydrate degradation in the lysosome.
|
-carbs synthesized and degraded in a step-wise manner
>lots of redundancy in protein degradation, but not carbo degradation >many disorders of carbo deg. -endopeptidases cleave protein seq in middle -exopeptidases cleave from N- or C-terminus (1 or 2 aa’s only) |
|
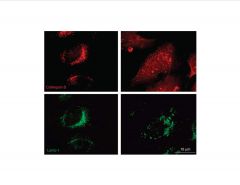
Analyze this data.
(Hydroxychloroquine added in panels on right) |
Hydroxychloroquine induces lysosomal membrane permeability (lmp).
-cathepsin beta=soluble, hydrolytic proteinase >prob not active at neutral pH >leaks out when treated with hydroxychloroquine (anti-malarial and anti-cancer Rx) -LAMP1=membrane protein (i.e. membrane still in tact) |
|
|
Describe lysosomal biogenesis.
|
Lysosomal proteins are synthesized on the rough endoplasmic reticulum and post-translationally modified in the RER and Golgi apparatus.
Lysosomal proteins are sorted in the Golgi apparatus to the late endosomes and lysosomes. This sorting requires AP-1 or AP-3 Soluble enzymes have a mannose 6-phosphate signal. Membrane proteins have a peptide signal. |
|
|
Describe the trafficking of lysosomal enzymes.
|
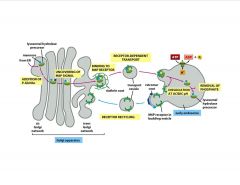
the M6P residues that direct proteins to lysosomes are generated in the cis-golgi by 2 resident enzymes:
1) N-acetyglucosamine (GlcNAc) phophotransferase transfers a phospho-GlcNAc to 1 or more mannose residues (added in ER). Only lysosomal enzymes have seq that are recognized and bound by this enzyme, so phospho-GlcNAc gp's are added only to lyso enzymes 2) After release from phosphotransferase, a phosphodiesterase removes GlcNAc gp, leaving phospho-mannose on lyso enzyme |
|
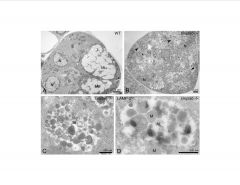
Analyze this data.
|
-mucolipidosismiss GlcNAC phosphotransferase enzyme (GNPtab)
>looking at exocrine glands >LAMP2=lysosomal protein -the soluble enzymes normally within the lysosomes are missing, and thus, fatty droplets accumulate and cause the vesicles to swell |
|
|
Compare the differences of presentation in an acinar/exocrine cell vs. a fibroblast that has lost Gnptab (GlcNAc) activity.
|
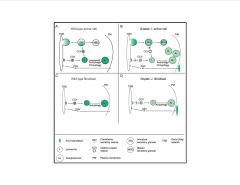
-exocrine cells have regulated secretion.
>they are normally secreters so to become constitutively active in the secretive pathway doesn't impact as much as fibroblasts which usually retain more of the enzyme |
|
|
What are the various delivery pathways to lysosomes?
|
Phagocytosis of extracellular organisms such as bacteria and yeast
Receptor-mediated endocytosis of extracellular hormones, proteins, macromolecules, lipid particles, and viruses Pinocytosis of extracellular proteins and glycosaminoglycans Crinophagy of intracellular secretory vesicles Chaperone-mediated transport of endogenous soluble proteins Microautophagy of endogenous soluble proteins, ribosomes, and glycogen Macroautophagy of macromolecules and organelles |
|
|
Describe the merger of the endocytic and biosynthetic pathways.
|
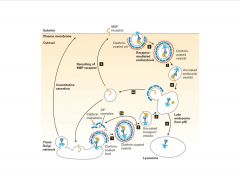
|
|
|
Compare micro- and macroautophagy.
|
-microautophagy= invagination of lysosome (direct route)
-macroautophagy=indirect route requiring fusion event |
|
|
What chaperone mediates selective transport of cytosolic proteins to the lysosome for degradation?
|
There exists a chaperone (hsc73) that mediates selective transport of cytosolic proteins into the lysosomes for degradation.
Lysosomal transport requires ATP, lysosomal membrane protein (LAMP2a), a hsc73 recognition domain within the target protein (KFERQ), and cytosolic and lysosomal hsc73. |
|
|
What are some properties of hsc73?
|
-soluble protein that traverses lysosomal membrane
>protein stretched to fit through channel >recognize KFERQ signal |
|
|
What is LAMP2? What disease does a defect cause?
|
-lysosomal membrane protein
-The results suggest that Lamp-2 is required for both chaperone-mediated autophagy and macroautophagy. -Danon disease is an X-linked disorder in which cells have a defective Lamp-2 This disease is clinically characterized by cardiomyopathy, myopathy, and mental retardation. |
|
|
Describe the breakdown and reformation of the nuclear envelope during mitosis.
|

|
|
|
Describe lamins and their function(s).
|

-lamins are class V IF's
>chromatin may attach directly or connect via other adapters |
|
|
Describe the structure and processing of lamins.
|
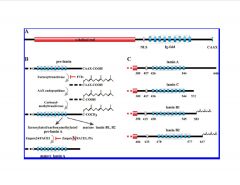
-farnesyl residue inserted at Cys of pre-lamin
-splicing leads to B1 and B2 -NLS, IgG repeats, CAAX box -FACE1 cleaves CAAX box to yield mature lamin -pre-lamin A, 646 recognized by FACE1 to cleave CAAX >mutation makes FACE1 unable to work >Hutchinson-Gilford progeria syndrome (G608) |
|
|
How are large proteins (>9 nm/60 kD) transported across the nucleus?
|

-sometimes requires adapter protein
|
|
|
Describe the mechanism of nuclear import.
|
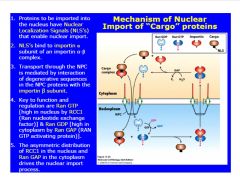
-mechanism of nuclear import:
>4 components: RAN (GTP or GDP), cargo, importin (receptor) -RCC!=Ran GEF >balance of GEF and GAP in cytoplasm vs. nucleoplasm drives import of materials (protein maintains tertiary structure...NPC accomodates protein shape) |
|
|
Describe export through the NPC.
|
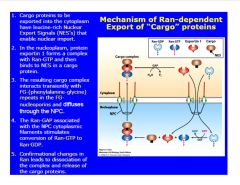
-must have export signal to be shuttled out of nucleus
>reverse process of import |
|
|
Describe Ran-independent export of mRNA's.
|
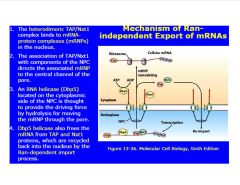
-Tap and Nxt1 recognize mature mRNA (binds close to polyA tail)
-Dbp5=RNA helicase >provides ATP to move from nucleus to cytoplasm |
|
|
What are the functions of the peroxisome?
|

|
|
|
Describe peroxisome biogenesis.
|

Pex 19=peroxisomal receptor
>facilitates association of other membrane proteins -Pex 5 recognize cargo protein with PTS-1, Pex7 recognizes PTS-2 >comprise mature peroxisome (with other factors) to recruit needed enzymes to core -mature peroxisome duplicates through fission using Pex11 |
|
|
Give an overview of the peroxisomal matrix protein import cascade.
|
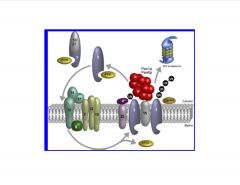
|
|
|
What are the key players in the peroxisomal matrix protein import cascade.
|
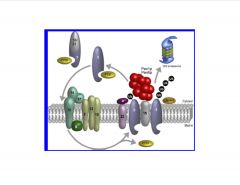
i)Cargo recognition in the cytosol and direction of the receptor-cargo complexes to the peroxisomal mem:
>2 major targeting signal Pex5p (perox. targeting signal 1 receptor) and PTSR2 have been id'd >The Pex5p signal is a carboxy-terminal tripep S-K-L-COO- >pro harboring the PTS1 signals are recognized by the soluble receptor Pex5p (PTSR1) >PTS2 signal pro's are recognized by Pex7p (PTSR2) ii)translocation of the receptor-cargo complex to the luminal site of the peroxisomal mem: >the receptor-cargo complexes assoc with the peroxi mem via the peroxi docking complex (Pex14p, 13p, and 17p) >the RING-finger domain containing peroxins (pex2p, 10p, and 12p) form the RING-finger complex, which is connected to the docking-complex via Pex8p. iii)disassembly of the receptor-cargo complex in the peroxi lumen: >receptor-cargo complex dissociates at the luminal site of the mem >integral PTSR1 is either monoubi by the E2 enzyme Pex4p or polyubi by Ubc4p or Ubc5p iv)return of the receptor to the cytosol: >the ATPase assoc with various cellular activities (AAA) peroxins Pex1p and 6p, which are anchored to the peroxi mem by Pex15p, dislocate the ubi Pex5p from the mem back to the cytosol >polyubi PTSR1 are degraded by the proteasome, whereas monoubi receptors are recycled for futher rounds of import (-import cascade: >Pex5 and 7 recognize cargo based on short seq >Pex 7 assoc w/Pex 18 and 21 to form complex >translocates to membrane andis recognized by ring-like structure composed of different Pex's (13, 14, and 17) >dissociates once inside >Pex 5 and 7 get exported back out >if mono-Ub, get moved out by Pex1p and 6p if poly-Ub=proteasomal degradative pathway) |
|
|
Describe what a mutation in Pex12 or 3 would look like.
|

-mutations involved in biogenesis or transport drastically effect the cell
-Pex12 mutation blocks import -Pex3 mutant blocks biogenesis >don't get inserted in membrane (remain diffusely in nucleus) |
|
|
What are the 2 types of peroxisomal diseases?
|

|
|
|
Describe the mitochondrial fusion proteins.
|

-all 3 fusion proteins are GTPase's, with TM domains and HR domains
>TM anchors to outer mito. membrane >HR anchors HR2 from neighboring mito. and pulls the 2 together OPA1 within the mito. becomes active once the 2 mito. are connected and allows fusion |
|
|
Describe the mitochondrial fusion process.
|
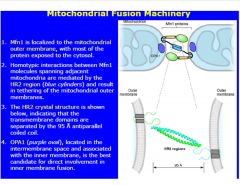
|
|
|
Describe mitochondrial fission proteins.
|

|
|
|
Describe mitochondrial fission.
|

Initial constriction of fission process is self-directed after maturation
>Drp1 recognizes this groove -once seqperated, 1 mito. has high nrg capacity and the other is low nrg >low nrg gets degraded |
|
|
Compare FA oxidation in the 2 organelles in which it occurs.
|

-acetyl CoA produced in FA oxidation in both peroxisomes and mito.
>acteyl CoAmust be transported from peroxisome to other organellles -e- transport system necessary for reducing O2 to water |
|
|
Describe the process of mitochondrial oxidation damage.
|

|
|
|
Describe the role of SOD in human disease.
|

|
|
|
What are some ways SOD mutants are implicated in motoneuron diseases?
|

mutations in SOD lead to aggregate formation (vs. SOD homodimers)
>aggregates complex with Bcl2 in mito. mem. >can cause mito. swelling, affect permeability, or release CytC (apoptotic) >can be tissue specific (may effect neuronal system more so than other organs) |
|
|
What organelle is often targeted by SOD1?
|
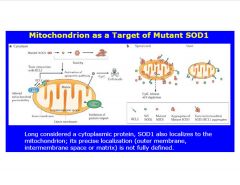
xs e- gradient produced by stroke (due to blockage of ATP production)
>produce ROS >p53 involved in trancscription of mitochondrial protein >when activated, can bind to mito. mem. and transcribe |
|
|
How do ROS initiate apoptosis?
|
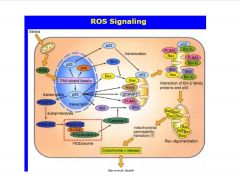
-mito-specific genes
>binds Bcl protein (in complex w/BAX) >BAX gets released (also, actively being transcribed >xs BAX forms a pore in mito. mem. though which Cyt C gets released and initiates apoptosis |
|
|
Compare peroxisomes and mitochondria.
|
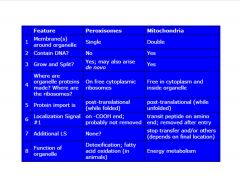
|
|
|
Describe the role of SOD in ALS.
|
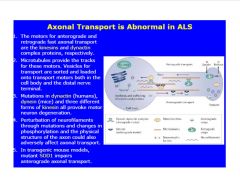
|