Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
141 Cards in this Set
- Front
- Back
|
When are poison exposures more common (by age group)?
|
- <6 years: 1 million reported
- 6-12 years: 140,000-150,000 - 13-19 years: 150,000-160,000 |
|
|
How common are ED visits and deaths for unintentional, non-fire related CO exposures?
|
- ED visits: 15,000
- Deaths: 500 |
|
|
How common are inborn errors of metabolism (per births)? How common are these amongst the single gene disorders?
|
- 1:1400 births
- 15% of single gene disorders |
|
|
What are the most common inherited metabolic disorders?
|
Lysosomal Storage Diseases (LSDS)
|
|
|
How are most Lysosomal Storage Diseases (LSDS) inherited? Exceptions?
|
- Most: Autosomal Recessive (eg, Niemann-Pick Disease, Type C)
- Exception: X-linked Recessive (eg, Fabry Disease and Hunter Syndrome (MPS II)) |
|
|
When should you suspect a metabolic disorder?
|
- When clinical presentation does not fit a medical textbook
- Patient does not respond to common treatment - Patient defies the "clinical rationale" |
|
|
What might cause a metabolic disorder to go unrecognized?
|
It may only become obvious during physical stress, fasting, or acute illness/injury
|
|
|
What are the possible implications of not recognizing a metabolic disorder and delaying treatment?
|
Can result in irreversible injury to the brain and major organs or death
|
|
|
What are the suspicious presentations for a metabolic disorder?
|
- Patient with unexplained lethargy, confusion, somnolence or coma (‘Do the Right Thing’)
- Patient with unexplained metabolic acidosis/alkalosis -- Patient with excessive lactate or ketosis - Patient with persistent or recurrent hypoglycemia - Patient with chronic and worsening symptoms (progression and regression are alarm signals) - Patient with unusual findings on MRI, EEG or pathology. - Patient with unusual combination of findings indicating a complex disease process or more than 1 etiology. |
|
|
What should you do for an unresponsive patient with unexplained lethargy, confusion, somnolence or coma?
|
- Physical exam and medical history (source?)
- Check glucose, ammonia, and pH (STAT) - Call your metabolic specialist, if available - Check electrolytes, CK, LFTs, lactate, urine analysis - Store a ‘critical sample’ (hypoglycemia) - Start treatment without delay (IV glucose) - Basic metabolic work-up: acylcarnitine profile, amino acid profile, urine organic acid profile - Don’t forget the alternatives (the 3 i’s: infection, intoxication and idiopathic) |
|
|
In an unresponsive patient with unexplained lethargy, confusion, somnolence or coma, what are the alternatives to a metabolic disorder?
|
3 "i's":
- Infection - Intoxication - Idiopathic |
|
|
What is the function of lysosomes?
|
- Intracellular digestive tract
- Acid hydrolases break down macromolecules |
|
|
What do Lysosomal Storage Diseases (LSD) result from?
|
Lack of any protein essential for normal function of lysosomes
|
|
|
What is the term for a neuronal storage disease?
|
Gangliosidose
|
|
|
What is the general cause of Gangliosidoses (type of neuronal storage disease)?
|
- Results from accumulation of gangliosides (abundant in brain) within neurons
- GM2 gangliosidoses are caused by deficiency of lysosomal enzymes |
|
|
What lysosomal enzymes can be deficient in Gangliosidoses? Specific diseases caused?
|
- Hexoaminidase A: Tay-Sachs Disease
- Hexosaminidase B: Sandhoff Disease - Activator Protein Deficiency: GM2 Gangliosidosis, variant AB |
|
|
What enzyme is deficient in Tay-Sachs Disease?
|
Hexoaminidase A
|
|
|
What enzyme is deficient in Sandhoff Disease?
|
Hexoaminidase B
|
|
|
What enzyme is deficient in GM2 Gangliosidosis, variant AB?
|
Activator Protein Deficiency
|
|
|
How does a lysosomal storage disorder compare to poisoning?
|
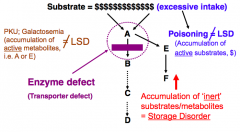
- Poisoning: accumulation of active substrates d/t excessive intake
- Lysosomal Storage Disorder: accumulation of inert substrates/metabolites (may be d/t enzyme defect) |
|
|
What happens to a cell when there is a lysosomal enzyme deficiency?
|
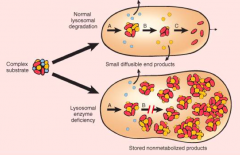
Cell swells because of storage of non-metabolized products
|
|
|
How do lysosomes normally work to break down macromolecules?
|
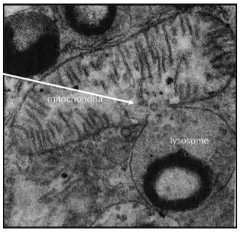
Lysosome releases digestive enzymes into a mitochondria to break down its macromolecules
|
|
|
What are the categories of Lysosomal Storage Disorders?
|
- Lysosome assembly (Golgi apparatus)
- Trafficking of lysosomal enzymes (glycosylation) - Enzyme deficiency (single gene defect) - Co-factor defect - Transporter defect - (Miner's Disease (Silicosis) and Asbestosis are not considered defects in lysosomal function) |
|
|
Tay-Sachs Disease has a high incidence in what population? Mutations are in a gene on what chromosome?
|
- High incidence in Ashekenazi Jews
- Gene on chromosome 15 (Hexoaminidase A), >100 mutations described |
|
|
How do you diagnose Tay-Sachs Disease?
|
Enzyme (Hexoaminidase A) assay of serum, WBC, or cultured fibroblasts
|
|
|
What are the clinical signs / symptoms of Tay-Sachs Disease?
|
- Normal at birth
- 6 months: psychomotor retardation evident - S/S may progress to blindness, motor incoordination, eventual flaccidity, mental deterioration, eventual decerebrate state - Cherry red spot in macula |
|
|
What is the prognosis for Tay-Sachs Disease?
|
Death by 2-3 years
|
|
|
What is necessary for effective Hexoaminidase A function (if not intact causes Tay-Sachs Disease)?
|
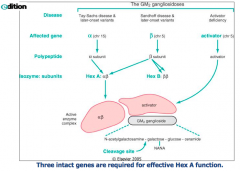
- α gene (chr 15)
- β gene (chr 5) - Activator gene (chr 5) |
|
|
What happens to the brain in Tay-Sachs Disease?
|
- Normal / little / big depending on duration
- If they survive >2 years, the brain will be large |
|
|
What are the light microscopic findings in Tay-Sachs Disease?
|
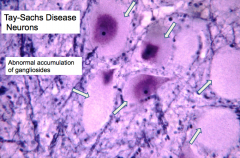
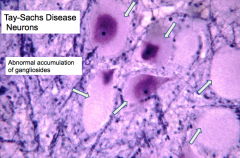
- Enlarged ballooned neurons filled w/ PAS positive material (stored gangliosides)
- Storage also in other brain cells (astrocytes and microglia) |
|
|
What are the electron microscopic findings in Tay-Sachs Disease?
|
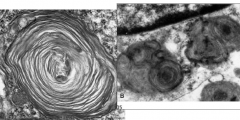
Membranous cytoplasmic bodies
|
|
|
How do you treat Tay-Sachs Disease?
|
Still in experimental stages:
- "Chaperone" proteins may help the α subunit to fold normally - Enzyme replacement therapy |
|
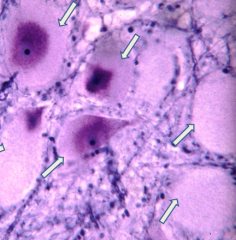
What do these arrows point out? Pathology?
|
Tay-Sachs Disease:
- Enlarged ballooned neurons filled w/ PAS positive material (stored gangliosides) - Storage also in other brain cells (astrocytes and microglia) |
|
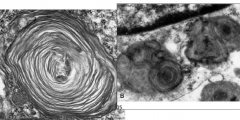
What are these swirls indicative of?
|
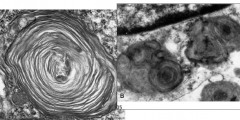
Tay-Sachs Disease:
- Membranous cytoplasmic bodies |
|
|
What is the other name for Krabbe's Disease?
|
Globoid Cell Leukodystrophy
|
|
|
What kind of disorder is Krabbe's Disease (Globoid Cell Leukodystrophy)? Cause?
|
- Lysosomal storage disease
- Autosomal recessive: gene on chr 14 - Deficiency of galactocerebroside-B-galactosidase |
|
|
What are the implications of a galactocerebroside-B-galactosidase deficiency?
|
Causes Krabbe's Disease (Globoid Cell Leukodystrophy)
- Enzyme deficiency (galactocerebroside-B-galactosidase) causes accumulation of toxic compound (psychosine) that injures oligodendrocytes - Galactocerebroside is a component of the myelin sheaths that accumulates in the "globoid cells" - Affects both CNS and PNS |
|
|
What accumulates in Krabbe's Disease (Globoid Cell Leukodystrophy)? Implications?
|
- Psychosine (toxic compound) injures oligodendrocytes
- Galactocerebroside accumulates in "Globoid cells" |
|
|
How do you diagnose Krabbe's Disease (Globoid Cell Leukodystrophy)?
|
Enzyme assay (galactocerebroside-B-galactosidase) of WBC or cultured fibroblasts
|
|
|
What is the clinical course of Krabbe's Disease (Globoid Cell Leukodystrophy)?
|
- Normal development at first
- Onset between 3-6 months ● Irritability, development ceases ● Deterioration of motor function: tonic spasms, eventual opisthotonic posture, myotonic jerking ● Optic atrophy, blindness ● CSF protein elevated |
|
|
What is the prognosis for Krabbe's Disease (Globoid Cell Leukodystrophy)? How do you treat?
|
- Death by approximately 2 years
- Tx: umbilical cord / BM transplant (in pre-symptomatic phase) |
|
|
What is the gross appearance of Krabbe's Disease (Globoid Cell Leukodystrophy)?
|
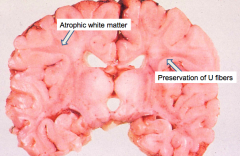
- Atrophic brain with firm white matter
- Atrophic white matter - Preservation of "U" fibers |
|
|
What is the light microscope appearance of Krabbe's Disease (Globoid Cell Leukodystrophy)?
|
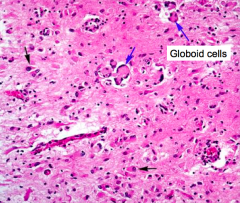
Presence of Globoid cells
- Loss of myelin - Accumulation of globoid macrophages, cluster around vessels - Severe astrocytosis - Decreased numbers of oligodendrocytes |
|
|
What is the electron microscope appearance of Krabbe's Disease (Globoid Cell Leukodystrophy)?
|
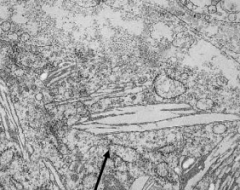
Globoid cells contain crystalloid straight or tubular profiles
|
|
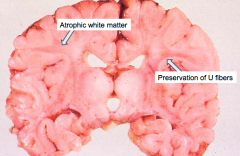
What are these gross findings signs of?
|
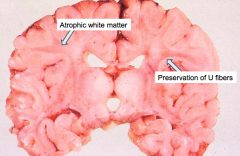
Krabbe's Disease (Globoid Cell Leukodystrophy)
|
|
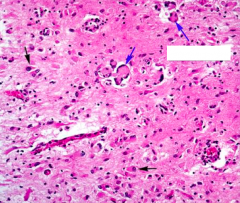
What do the arrows point at? Pathology?
|
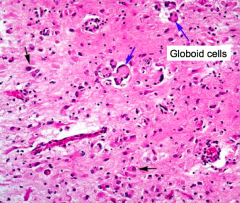
- Globoid Cells
- Sign of Krabbe's Disease (Globoid Cell Leukodystrophy) |
|
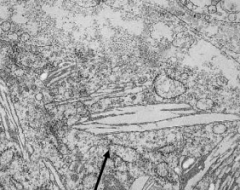
What do the arrows point at? Pathology?
|
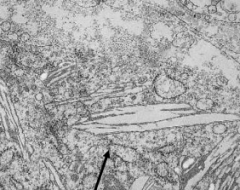
- Globoid Cells contain crystalloid STRAIGHT or tubular profiles
- Sign of Krabbe's Disease (Globoid Cell Leukodystrophy) |
|
|
What lysosomal metabolic disorder is caused by a deficiency of Aryl Sulfatase A?
|
Metachromatic Leukodystrophy
|
|
|
What kind of disease is Metachromatic Leukodystrophy? Cause?
|
- Lysosomal storage disease
- Autosomal recessive inheritance of gene on chr 22 - Deficiency of aryl sulfatase A |
|
|
What happens in Metachromatic Leukodystrophy?
|
- Metachromatic lipids (sulfatides) accumulate in brain, peripheral nerves, and kidney (d/t deficiency of aryl sulfatase A)
- Sulfatide accumulation leads to break down of myelin |
|
|
How do you diagnose Metachromatic Leukodystrophy?
|
Demonstrate Aryl Sulfatase A deficiency in urine, WBC, or fibroblasts
|
|
|
What are the implications of sulfatide accumulation in Metachromatic Leukodystrophy?
|
Leads to break down of myelin
|
|
|
What are the clinical signs/symptoms of Metachromatic Leukodystrophy?
|
Childhood: presents with gait disorder and motor symptoms
- Late infantile presentation (most common) - Intermediate - Juvenile Adult: usually presents with psychosis and cognitive impairment, eventual motor symptoms |
|
|
What is the prognosis and treatment for Metachromatic Leukodystrophy?
|
- Death in 5-10 years (in childhood type)
- Treatment: BM stem cell transplant (before symptoms appear) |
|
|
What is the gross appearance of Metachromatic Leukodystrophy?
|
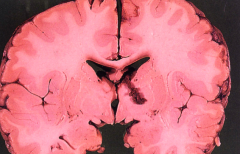
- Brain is externally normal
- White matter is very firm - Marked loss of myelin - Preservation of "U" fibers (subcortical fibers) |
|
|
What is the microscopic appearance of Metachromatic Leukodystrophy?
|
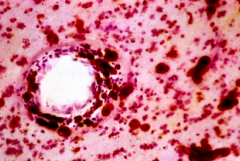
Metachromasia of white matter deposits (brown staining) - via acidified CRESYL VIOLET stain
|
|
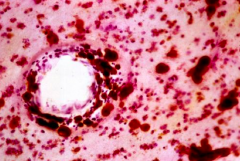
What are this finding a sign of?
|
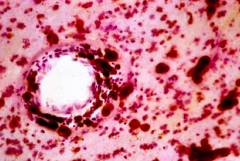
Metachromatic Leukodystrophy
- Brown staining via acidified cresyl violet stain - Metachromasia of white matter deposits |
|
|
What is the other name for Adrenoleukodystrophy?
|
Schilder's Disease
|
|
|
What is the cause of Adrenoleukodystrophy (Schilder's Disease)?
|
- Decreased activity of very long chain fatty acyl-CoA synthetase (in peroxisomes)
- X-linked (classic form) |
|
|
What is the function of peroxisomes?
|
Cytoplasmic spherical "microbodies"
- Contain catalase - Involved in fatty acid β-oxidation (and more) |
|
|
What are the implications of decreased activity of very long chain fatty acyl-CoA synthetase (in peroxisomes) in Adrenoleukodystrophy (Schilder's Disease)?
|
Excess of very long chain fatty acid esters in plasma, cultured fibroblasts, and affected organs (CNS, PNS, adrenal glands)
|
|
|
What is the classic presentation of Adrenoleukodystrophy (Schilder's Disease)?
|
- Onset between 5-9 years or 11-21 years
- Neuro S/S: dementia, visual/hearing loss, seizures - Adrenal insufficiency follows neuro s/s |
|
|
What is the non-classic presentation of Adrenoleukodystrophy (Schilder's Disease)?
|
Adrenomyeloneuropathy
- Occurs in adult (20-30 years) - Slowly progressive leg clumsiness/stiffness - Eventual spastic paraplegia - Adrenal insufficiency may precede neuro s/s |
|
|
How do the classic and adrenomyeloneuropathy forms of Adrenoleukodystrophy (Schilder's Disease) differ in terms of age of onset, symptoms, and presence of adrenal insufficiency?
|
Classic:
- Onset: 5-9 years or 11-21 years - S/S: dementia, visual/hearing loss, seizures - Adrenal insufficiency: follows neuro s/s Adrenomyeloneuropathy: - Onset: adults (20-30 years) - S/S: slowly progressive leg clumsiness/stiffness, eventual spastic paraplegia - Adrenal Insufficiency: may precede neuro s/s |
|
|
What is the gross appearance of Adrenoleukodystrophy (Schilder's Disease)?
|
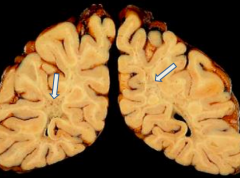
- Gray discoloration of white matter; marked firmness
- Severe demyelination - "U" fiber preservation |
|
|
What is the microscopic appearance of Adrenoleukodystrophy (Schilder's Disease)?
|
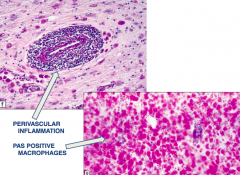
- Perivascular inflammation
- PAS positive macrophages |
|
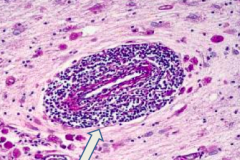
What is the arrow pointing at? Pathology?
|
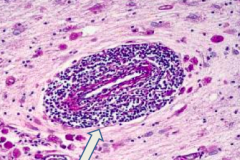
Perivascular inflammation: sign of Adrenoleukodystrophy (Schilder's Disease)
|
|
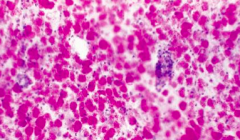
What is the arrow pointing at? Pathology?
|
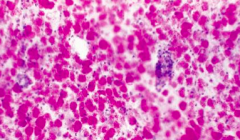
PAS positive macrophages: sign of Adrenoleukodystrophy (Schilder's Disease)
|
|
|
What can occur as a complication of severe liver disease or chronic portocaval shunting?
|
Hepatic Encephalopathy
|
|
|
What causes Hepatic Encephalopathy?
|
Partially related to hyperammonemia
|
|
|
What are the early and late features of Hepatic Encephalopathy? Other symptoms?
|
- Early: inattentiveness and short term memory impairment
- Late: confusion, asterixis (flapping tremor of outstretched hands), drowsiness, stupor, and coma - Patients may also have foul breath (fetor hepaticus), hyperventilation, gait disturbances, and choroathetosis |
|
|
What are the signs of Hepatic Encephalopathy on MRI?
|
- Increased T1 signal in the globus pallidus, subthalamus, and midbrain
- Cortical edema |
|
|
What is the prognosis and treatment for Hepatic Encephalopathy?
|
- Acute Hepatic Encephalopathy can be rapidly fatal
- Chronic or repeated episodes can lead to permanent and progressive neuropsychiatric disturbances - Chronic Hepatic Encephalopathy / Myelopathy can be ameliorated by a liver transplantation |
|
|
What is the microscopic appearance of Hepatic Encephalopathy?
|
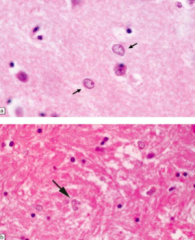
Alzheimer Type II Astrocytes
|
|
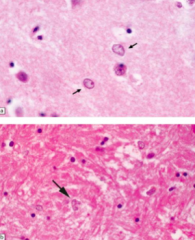
What do these arrows point out? Pathology?
|
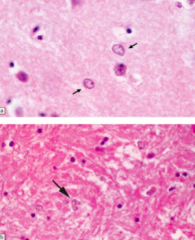
- Alzheimer Type II Astrocytes
- Sign of Hepatic Encephalopathy |
|
|
What can cause hypoglycemia?
|
- Insufficient food intake
- Systemic diseases (primary hyperinsulinism, severe liver disease, or adrenal insufficiency) - Exposure to drugs that cause hypoglycemia (insulin) |
|
|
How do patients with hypoglycemia present?
|
- Headache
- Confusion - Irritability - Incoordination - Lethargy that may lead to stupor and coma |
|
|
What are the MRI findings of hypoglycemia?
|
Signal changes in the temporal, occipital, and insular cortices, hippocampus, and basal ganglia, often with thalamic sparing
|
|
|
Which parts of the brain are particularly vulnerable to hypoglycemia? Changes?
|
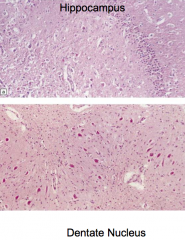
Red neurons
- Lamina 3, 5, and 6 - Putamen - Caudate nucleus - Dentate nucleus |
|
|
How do you treat hypoglycemia?
|
Depends on cause:
- Restoration of glucose for exogenous causes - Removal of endogenous causes (liver, pancreatic, or adrenal tumors) |
|
|
What organs are affected by mitochondrial diseases?
|
Can cause a broad variety of clinical issues, involving numerous organ systems:
- Common: brain and muscle involvement - Often: GI tract, heart, and/or peripheral nerves |
|
|
How are mitochondrial diseases inherited?
|
Maternal inheritance (mitochondrial)
|
|
|
Where are mitochondrial proteins encoded?
|
With the mitochondrial AND nuclear genome (complicates diagnosis)
|
|
|
How do you diagnose mitochondrial diseases?
|
- Based on a battery of clinical, imaging, pathoogical, and molecular assays
- Highly variable by institution |
|
|
Are there specific mutations associated with mitochondrial diseases?
|
- Specific mutations in the mitochondrial genome are associated with specific diseases
- Most patients with mitochondrial disease do not fit these specific syndromes - There are also ~1000 nuclear genes that can contribute to the mitochondrial phenotype |
|
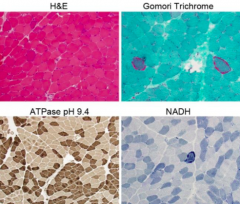
What do these stains of muscle tissue indicate?
|
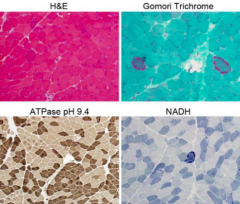
Mitochondrial Myopathy
|
|
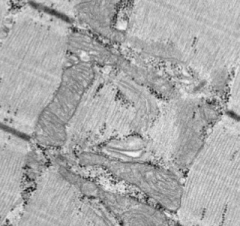
Is this EM normal or abnormal? If abnormal, what does it indicate?
|
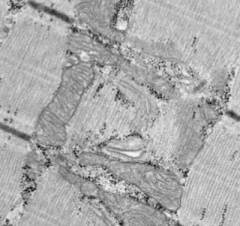
Normal - no mitochondrial myopathy
|
|
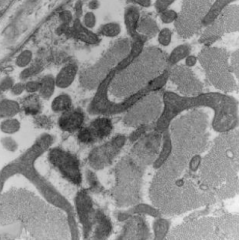
Is this EM normal or abnormal? If abnormal, what does it indicate?
|
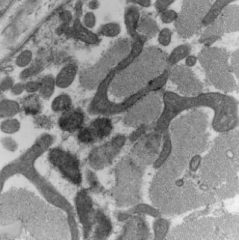
Abnormal - Mitochondrial Myopathy
|
|
|
What are the mitochondrial syndromes?
|
- Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS)
- Myoclonic Epilepsy with Ragged Red Fibers (MERRF) - Kears-Sayre Syndrome (KSS) - Leigh's Disease |
|
|
What is MELAS? What kind of disease? Cause?
|
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS)
- Mitochondrial syndrome - Usually caused by heteroplasmic point mutations in mt-tRNA(Leu) |
|
|
What is MERRF? What kind of disease? Cause?
|
Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
- Mitochondrial Syndrome - Usually caused by heteroplasmic point mutations in mt-tRNA(Lys) |
|
|
What is KSS? What kind of disease? Cause?
|
Kears-Sayre Syndrome (KSS)
- Mitochondrial Syndrome - Usually caused by large single mtDNA mutations - Causes pigmentary retinopathy and ophthalmoplegia before 20 years of age |
|
|
What is Leigh's Disease? What kind of disease? Cause?
|
- Mitochondrial Syndrome
- Caused by a nuclear mutation (and mitochondrial DNA have been reported) - Causes enzyme deficiency in pathway converting pyruvate to ATP - Decreased activity of cytochrome C oxidase - Autosomal recessive |
|
|
What is caused by heteroplasmic point mutations in mt-tRNA(Leu)?
|
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS)
- Mitochondrial syndrome |
|
|
What is caused by heteroplasmic point mutations in mt-tRNA(Lys)?
|
Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
- Mitochondrial Syndrome |
|
|
What is caused by pigmentary retinopathy and ophthalmoplegia before 20 years of age?
|
Kears-Sayre Syndrome (KSS)
- Mitochondrial Syndrome |
|
|
Which mitochondrial syndrome is caused by a nuclear mutation? How is it inherited?
|
Leigh's Disease - autosomal recessive
|
|
|
What are the signs/symptoms of Leigh's Disease? Prognosis?
|
- Lactic acidemia
- Arrest of development, hypotonia, seizures, extraocular palsies - Death between 1 and 2 years |
|
|
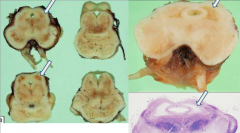
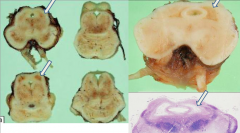
What is the gross appearance of Leigh's Disease?
|
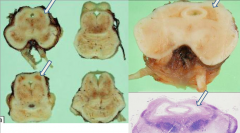
Periventricular gray matter tissue destroyed (around cerebral aqueduct and third ventricle)
|
|
|
What is the microscopic appearance of Leigh's Disease?
|
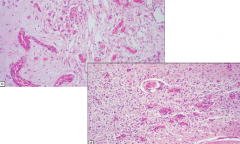
Spongiform appearance and vascular proliferation
|
|
What is this pathology?
|
Leigh's Disease
- Periventricular gray matter tissue destroyed (around cerebral aqueduct and third ventricle) |
|
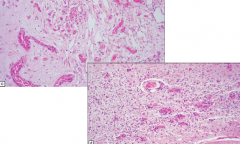
What is this pathology?
|
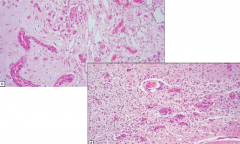
Leigh's Disease
- Spongiform appearance and vascular proliferation |
|
|
Which vitamin deficiency is predominantly seen in malnourished chronic alcoholics?
|
Thiamine (Vitamin B1)
|
|
|
What can cause Thiamine (Vitamin B1) Deficiency?
|
* Predominantly seen in malnourished chronic alcoholics
- Starvation diets - Hemodialysis - Gastric stapling - Extensive GI surgery - Hyperalimentation without thiamine supplementation |
|
|
What syndromes are caused by Thiamine (Vitamin B1) Deficiency?
|
- Wernicke Encephalopathy
- Korsakoff Syndrome |
|
|
What is the clinical presentation of Wernicke Encephalopathy? Cause?
|
- Ophthalmoplegia and nystagmus
- Ataxia - Confusion, disorientation, eventual coma - Caused by Thiamine (Vitamin B1) deficiency |
|
|
What is the gross appearance of Wernicke Encephalopathy?
|
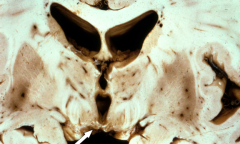
- Lesions are in mammillary bodies, dorsomedial thalamus, and around 3rd/4th ventricles
- Acutely: gray-brown discoloration with petechial hemorrhages - Chronically: atrophy and discoloration of mammillary bodies |
|
|
What is the microscopic appearance of Wernicke Encephalopathy?
|
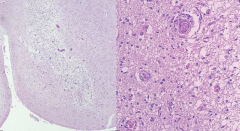
- Pallor
- Myelin loss - Prominent vessels - Macrophages - Relative preservation of neurons |
|
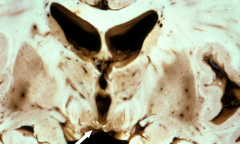
What is this pathology?
|
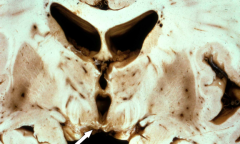
Wernicke Encephalopathy
- Lesions are in mammillary bodies, dorsomedial thalamus, and around 3rd/4th ventricles - Acutely: gray-brown discoloration with petechial hemorrhages - Chronically: atrophy and discoloration of mammillary bodies |
|
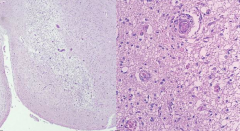
What is this pathology?
|
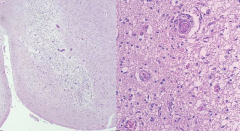
Wernicke Encephalopathy
- Pallor - Myelin loss - Prominent vessels - Macrophages - Relative preservation of neurons |
|
|
What are the clinical signs/symptoms of Korsakoff Psychosis?
|
- Loss of anterograde episodic memory
- Confabulation - Preserved intelligence and learned behavior |
|
|
What causes Korsakoff Psychosis?
|
- Hypothesized to result from repeated episodes of Wernicke's encephalopathy
- Damage to medial dorsal nucleus of thalamus |
|
|
How does Wernicke's Encephlopathy compare to Korsakoff Psychosis?
|
- Korsakoff Psychosis may result from repeated episodes of Wernicke's encephalopathy
- They have no distinct pathology |
|
|
What vitamin deficiency leads to subacute combined degeneration of the spinal cord?
|
Vitamin B12 (Cobalamin)
|
|
|
What could cause Vitamin B12 (Cobalamin) deficiency?
|
Pernicious Anemia (40% of untreated patients)
|
|
|
What are the clinical signs/symptoms of Vitamin B12 (Cobalamin) deficiency?
|
Subacute combined degeneration of the spinal cor:
- Ataxia - Romberg - Spasticity - Decreased reflexes - Mental status changes - |
|
|
What are the pathological signs of Vitamin B12 (Cobalamin) deficiency?
|
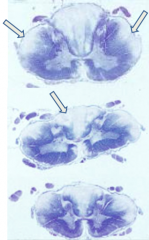
- CNS and PNS both involved
- Spinal cord: anterior and lateral corticospinal tracts and posterior columns are vacuolated and demyelinated - May have secondary axonal degeneration |
|
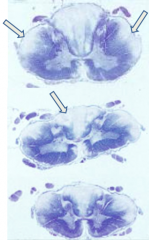
What pathology do the arrows show?
|
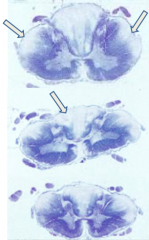
Vitamin B12 (Cobalamin) Deficiency: subacute combined degeneration of the spinal cord (anterior and lateral corticospinal tracts and posterior columns are vacuolated and demyelinated)
|
|
|
What is the effect of Carbon Monoxide?
|
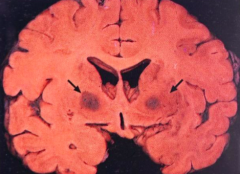
- Irreversibly binds to Hemoglobin, displacing Oxygen
- Binds to areas rich in iron (globus pallidus, substantia nigra) causing necrosis |
|
|
What parts of the brain are particularly affected by Carbon Monoxide poisoning?
|
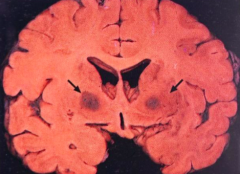
- Globus Pallidus
- Substantia Nigra |
|
|
How does Carbon Monoxide affect the brain?
|
- Binds to areas rich in iron (globus pallidus and substantia nigra) causing necrosis
- Degeneration of white matter |
|
|
Besides hemoglobin and brain tissue, how does CO affect the body?
|
- Usually accompanied by hypotension and ischemia
- Motor, cognitive, psychiatric, and parkinsonian signs/symptoms |
|
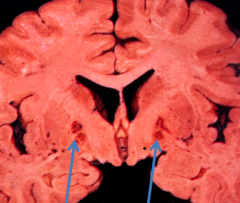
What do the arrows point out? Pathology?
|
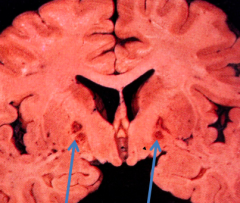
Bilateral pallidal cavitation - sign of chronic carbon monoxide poisoning
|
|
|
What are the signs/symptoms of chronic ethanol toxicity (alcoholism)?
|
- Truncal ataxia
- Nystagmus - Limb incoordination |
|
|
What are the gross findings of chronic ethanol toxicity (alcoholism)?
|
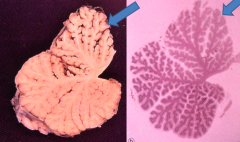
Cerebellar degeneration
- Atrophy, especially of anterior superior vermis - Dropout of Purkinje cells , internal granular cells, and astrocytosis |
|
|
What are the clinical signs / symptoms of Fetal Alcohol Syndrome?
|
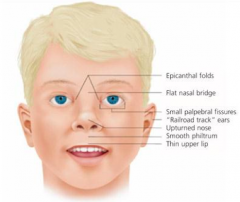
- Growth retardation
- Facial deformities: short palpebral fissure, epicanthal folds, thin upper limb, growth retardation of jaw - Cardiac defects: atrial septal defect - Delayed development and mental deficiency |
|
|
What are the pathological findings of Fetal Alcohol Syndrome?
|
- Microcephaly
- Cerebellar dysgenesis - Heterotropic neurons |
|
|
What is the hypothesized pathology responsible for Fetal Alcohol Syndrome?
|
Hypothesized that acetaldehyde crosses the placenta and damages fetal brain
|
|
|
When do the effects of radiation occur?
|
Months to years after exposure
|
|
|
What are the clinical symptoms of radiation toxicity?
|
Symptoms of a mass lesion
|
|
|
What pathology does radiation toxicity cause?
|
- Large areas of coagulative necrosis, primarily in white matter
- Vessels have marked thickening of the walls - Induction of neoplasms (meningiomas, sarcomas, gliomas) years after treatment |
|
|
What drugs are toxic to the CNS?
|
- Methotrexate
- Vincristine - Phenytoin - Cocaine - Amphetamine |
|
|
What are the drug toxicity effects of Methotrexate? Route of administration?
|
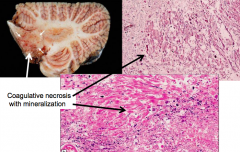
Intrathecal or intraventricular administration in combination with radiation may produce:
- Disseminated necrotizing leukoencephalopathy - Particularly around ventricles and deep white matter - Coagulative necrosis with axonal loss and mineralization |
|
|
What are the drug toxicity effects of Vincristine? Route of administration?
|
- Oral admin: sensory neuropathy
- Intrathecal admin: axonal swelling |
|
|
What are the drug toxicity effects of Phenytoin? Route of administration?
|
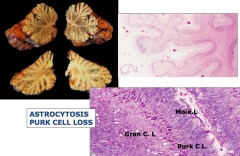
- Ataxia, nystagmus, slurred speech, and sensory neuropathy
- Atrophy of cerebellar vermis and loss of Purkinje cells and granule cells - Oral admin |
|
|
What are the drug toxicity effects of Cocaine?
|
- Seizures, strokes, hemorrhages
- Infarcts and hemorrhages d/t vasospasm, emboli, hypercoagulability, hypotension, and drug contaminants - Occasionally vasculitis (allergy?) |
|
|
What are the drug toxicity effects of Amphetamine?
|
- Infarcts and hemorrhages
- Attributed to vasculitis and hypertension |
|
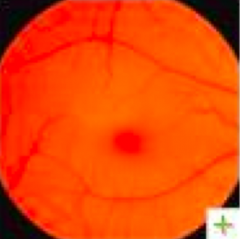
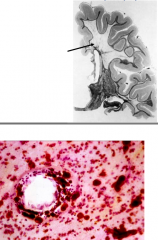
What is the differential diagnosis for "cherry red spots"? Cause?
|
Caused by lipid storage in the retinal layer of the fovea centralis (macula is transparent)
- Acute ophthalmic artery obstruction (typically unilateral) - GM1 gangliosidosis - Tay-Sachs/Sandhoff disease - Nieman-Pick disease - Mucolipidosis |
|
|
Which of the following is characteristic of hepatic encephalopathy?
a) Bilateral necrosis of the basal ganglia b) Cerebellar atrophy c) Demyelination with sparing of U fibers d) The presence of Alzheimer type II astrocytes e) The presence of globoid neurons |
The presence of Alzheimer type II astrocytes
|
|
|
Which of the following is NOT a typical clinical finding in fetal alcohol syndrome?
a) Catch-up in growth after being born with intrauterine growth retardation b) Behavioral abnormalities (i.e. ADHD) c) Epicanthal folds and flat nasal bridge d) Flat philtrum and thin upper lip e) Hypoplasia of the corpus callosum |
Catch-up in growth after being born with intrauterine growth retardation
|
|
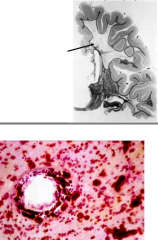
Which disease process is most likely to produce the pathology seen here?
a) Adrenoleukodystrophy b) Krabbe’s globoid cell leukodystrophy c) Leigh’s disease d) Metachromatic Leukodystrophy e) Tay Sachs disease |
Metachromatic Leukodystrophy
|