![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
133 Cards in this Set
- Front
- Back
|
New generation of drugs |
SSRIs, designer drugs, cognitive enhancers |
|
|
drug action vs drug effect |
drug vs receptor behaviour |
|
|
pharmacokinetics |
absorp, transp, metab, elim |
|
|
bioavailability |
how much drug is in the brain |
|
|
differences in bioavailability |
absorption (oral slow absorp & peak; nasal fast absorp & peak) |
|
|
cell membrane |
phospholipid bilayer |
|
|
polar molecule |
charged/water soluble |
|
|
nonpolar molecule |
uncharged/fat soluble |
|
|
5 transmembrane processes (FAPPP) |
passive diffusion: uncharged, pass through filtration: filter through cracks (semi permeable); must be small molecule active transport: Na K pumps passive transport: concentration gradient Phago/pinocytosis: invaginates molecule/liquid |
|
|
drug half life |
the time it takes for a drug to reach its peak concentration & be reduced/metabolised to half of the peak level |
|
|
steady state plasma level |
plasma concentration of drug wanted - continuous dose = build up T 1/2 - continuous level of drug in system |
|
|
partition coefficient |
how fat soluble a drug is - mix drug with water and oil = affinity for oil or water determined by pH |
|
|
pKa |
pH measure at which the drug will be 50% water soluble & 50% fat soluble |
|
|
ampoteric |
2 pKas |
|
|
Weak acid in acidic or basic environment |
tries to get rid of ion -less ionized/uncharged = fat soluble - ionized/charged = water soluble |
|
|
Weak base in basic or acidic environment |
tries to gain ion -less ionized/uncharged = fat soluble - ionized/charged = water soluble |
|
|
Work: weak acid; pKa 3.5; how well absorbed in stomach 2 pH & intestine 6 pH & blood 7.4 pH |
Stomach: mostly absorbed Intestines: less Blood: least |
|
|
distribution |
drug circulates in the blood |
|
|
2 Things affect distribution |
1. Depot binding: drug binds to other molecule and store in other places (fat) - more body fat = difficulty circulating 2. Water/Fat solubility of drug: fat soluble pass through vein membranes - carriers (albumin) to transport through circulatory system |
|
|
3 ways How drugs cross BBB |
fat soluble drugs transporter < 2000 mw = filter through |
|
|
BBB set up |
no immune system = hxc barrier made of neuronal & glial cells |
|
|
area without BBB - why |
area postrema detects harmful bodies & gives response (empty body) |
|
|
Placental barrier protection |
metabolites for cortisol fat soluble drugs pass through |
|
|
Where drugs metabolised |
liver by enzymes; enzymes also everywhere in cells = metabolised all over |
|
|
Primary method of metabolism |
liver; first pass metabolism; via hepatic portal venus system blood vessels from stomach and intestines pass through liver- after circulation, continuous liver metabolism |
|
|
biotransformation/metabolic clearance |
process liver breaks down drugs |
|
|
p450 enzymes role |
breakdown drugs 2 ways phase 1: additive process phase 2: make new componds |
|
|
types of drug receptors |
1. ionotropic/ligand gated ion channels (instantaneous) 2. metabotropic/gpcr (g protein channel receptor) (slower) 3. steroidal |
|
|
ionotropic works how |
resting state: channel blocked active state: ligand binds; conformational/shape change, channel opens, ion flow depolarizes, reaction |
|
|
metabotropic works how |
7 transmembrane structures (receptor domain in cytoplasm & cell), g protein (alpha, beta and gamma subunits) - Ligan binds to outside membrane of receptor, cause conformational change, inside receptor bump into g protein= activation - g protein separates = catalytic (activates second messengers) |
|
|
point of 2nd messenger cascade & how works |
amplify message - 1 receptor activates thousands of messengers (takes a while) -g protein becomes catalytic - activates second messengers - they actiavte other things in cell |
|
|
steroid/nuclear receptor |
steroids pass through membrane, binds to receptor in cell, 2 other receptors come and pair up (dimerizing), float into nucleus, bind to DNA, act as transcription factor BUT metabotropic estrogen receptor |
|
|
dimerizing |
two steroid receptors join |
|
|
pharmacodynamics |
relationship between drug and receptor (interaction) |
|
|
common elimination |
renal |
|
|
p450 enzymes individual differences |
sex modified induction |
|
|
induction |
more exposure to biotransformation = enzyme upregulates/gets better at drug breakdown = less drug to brain, takes more drug in system pharmacokinetic tolerance |
|
|
2 types of drug tolerance |
changes to receptors changes to liver enzymes |
|
|
drug-drug interaction: cross induction |
drug metabolised by same p450 enzyme as current drug taking -still some tolerance due to induction even though drug never taken before -ex: questions about general anesthesia |
|
|
opposite of cross induction |
grapefruit overhwelms certain p450 enzyme, drugs not broken down - builds up to dangerous levels in circulatory |
|
|
bio-activation |
liver metabolism activates the drug acetyl in front of calicetic acid |
|
|
main purpose of first pass metabolism |
make drug metabolites more water soluble - longer in circulatory system & so most likely eliminated |
|
|
Ligand |
bind to receptor endogenous: from inside exogenous: from outside |
|
|
Agonist |
drug ligand does same as endogenous ligand |
|
|
Allosteric binding |
drug ligand binds to site different than endogenous ligand binds |
|
|
Law of mass action |
L + R = LR* - Ligand + Receptor = Ligand*Receptor drug action - ligand pops in and out of binding with receptor - the more drug concentration = more binding time |
|
|
Weak bonds |
ionic, reversible, vanderwaals |
|
|
strong bonds |
covalent, irreversible bonds |
|
|
affinity |
how well drug ligand binds with specific receptor; dissociation with/without receptor - KD (dissociation constant) |
|
|
Antagonist |
drug ligand binds to compete with/prevent other molecules from binding (allosteric or active site) - does not activate/cause conformational change in receptor - have stronger bonds |
|
|
All receptors are: |
proteins |
|
|
Characteristics of Anatagonist active binding site |
reversible weak bond competitive more ligand/agonist = compete with antagonist |
|
|
Characteristics of Antagonist allosteric binding site |
irreversible - strong bond non-competitive |
|
|
most ligand bonds with receptors are: |
weak bonds except allosteric antagonists |
|
|
ionotropic properties |
5 subunits - 5 different proteins that come together to make a central channel - binding usually occurs on alpha - a lot of diversity (different combs of subunits) - determines how receptor will react to drug (different types of similar receptor) |
|
|
Inverse agonist |
effect on receptor (conformational change) = opposite effect of endogenous ligand |
|
|
partial agonist |
when saturate receptor, doesnt activate to maximal effect (conformational change not as big) |
|
|
full agonist |
when saturate receptor, activate them to maximal effect |
|
|
allosteric agonist |
enhance endogenous ligand by binding to alternative site |
|
|
allosteric antagonist |
inhibit activity of endogenous ligand/agonist, but doesnt prevent binding |
|
|
pharmacodynamic sensitization |
opposite of tolerance: receptors more sensitive to ligand - take antagonist chronically = neuron produces more receptors at site (excessive) - when antagonist removed =sensitized response - more receptors = more effect when agonist/ligand is in brain |
|
|
3 mechanisms of pharmacological tolerance |
1. de-sensitization 2. sequestration/endocytosis 4. gene down regulation |
|
|
de-sensitization |
phosphorylate receptor - less binding (fast) |
|
|
Sequestration |
flips receptor upside down (wont bind) or membrane invaginates receptor & internalizes them (recycle & elimination) |
|
|
Gene down regulation |
stop gene that transcribes the receptor (mRNA for that receptor), no new receptor being made while old removed |
|
|
pharmacodynamic tolerance |
too much ligand = down regulates receptors to maintain equilibrium and reduce effect - once ligand removed, receptors still low = withdrawal effect |
|
|
Saturation binding curve |
drugs affinity to bind with tissue - add increasing amount of drug to tissue - rinse excess drug off, rest bound |
|
|
Draw & explain saturation binding curves |
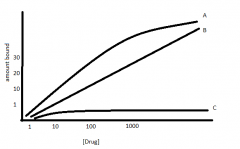
A - Total binding: amount of ligand bound specific receptor & non specific B - Non specific binding: another drug with same receptor saturate. Our drug cant get onto receptor, so can only bind to non-specific sites (linear) C - Receptor bound: A - B |
|
|
Bmax |
maximal binding (maximal amount of ligand bound to receptor) - where C (receptor bound ligand) plateaus & corresponding Y-axis - dose cannot increase |
|
|
Dissociation constant/KD/KD50 |
concentration at which the ligand it 50% bound to its receptor - Bmax/2 & corresponding mg on Y-axis - always a concentration - number used for affinity |
|
|
Lower KD means.. |
higher affinity for receptor (less amount of drug to bind 50% of receptors) |
|
|
3 pillars of drug potency |
1. Accessibility: Can cross BBB better (pKa) 2. Affinity: time spent stuck to receptor (KD50) 3. Efficacy: percentage of maximal change to receptor (EMax) |
|
|
Scatchard Plot why & data |
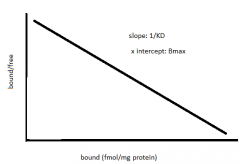
- slope = -1/KD50 - x intercept = Bmax - if not perfect straight line = multiple receptor binding sites |
|
|
Log scale |
convert numbers to log10 = sigmoidal curve |
|
|
types of dose response curves |
-graded dose response -quantal dose responsr |
|
|
graded dose response |
individual estimates - Emax: maximal effect: when curve asymptotes/plateaus - EC50: concentration of drug where we see 50% of the maximal effects |
|
|
drug efficacy |
- % of maximal change - how much can a drug do maximally compared to another drug - Emax- from drug response curve |
|
|
quantal dose response curve |

-population estimate - LD50 - TD50 - ED50 |
|
|
Potency measured by |
EC50 - graded dose response curve - smaller EC50 = higher potency (take less drug to get half of maximal efficacy) |
|
|
LD50 |
lethal dose 50: 50% population dies |
|
|
TD50 |
Toxic dose 50: 50% population gets side effects |
|
|
ED50 |
effective dose 50: 50% population gets wanted effect |
|
|
Therapeutic Index |
Ratio of drug dose that causes therapeutic effect:toxic effect ED50/LD50 OR ED50/TD50 (more conservative) large value = narrow range of safety |
|
|
Brake Research: Hippocampus memory type |
place learning more |
|
|
Brake Research: Dorsal Stratum memory type |
Response learning more |
|
|
Brake Research: New metabotropic estrogen receptor where? |
synapse in PFC, not NA |
|
|
How neuron fires |
all or none model - neuron fires or not, no inbetween |
|
|
parts of neuron |
soma, dendrites, axon, axon hillock, nodes of ranvier |
|
|
Neuron voting to fire or not |
other neurons attached, yes or no (excitatory or inhibitory) - soma sums the amount of + or -, |
|
|
can only fire or not = but different types of firing |
slow, fast, burst firing |
|
|
spatial summation |
location of input critical - excitatory input closer to membrane closer to axon hillock has greater influence |
|
|
temporal summation |
more rapid firing from other neuron onto soma has more influence on soma fires/not |
|
|
2 things affect neuron firing |
temporal and spatial summation |
|
|
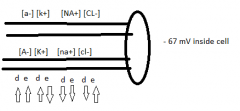
2 properties of resting potential |
diffusion: flow away from high concentration, towards low - electrostatic force: opposites attract, similar repel |
|
|
cations |
positive net charge |
|
|
anions |
negative net charge |
|
|
|
|
|
K pump |
out of cell - diffusion inside cell - electrostatic force constantly flowing back and forth until equilibrium ~ 80mV |
|
|
equilibrium potential |
the charge charge of membrane that ion no longer flows in or out |
|
|
Na K pumps |
use active transport to keep equilibrium since cell membrane is is leaky (keep concentration gradient) 3 Na for 2 K |
|
|
resting state charge of cell |
-65 |
|
|
Voltage gated Na channels open |
voltage gated Na channel opens @ -40 mV - Na floods in because diffusion & electrostatic - increase charge of membrane super fast- up to +40 mV |
|
|
Excess K CNS |
BBB stops excess K to enter brain - if enter, glial astrocytes soak up K |
|
|
Excess K PNS |
no protection = murder |
|
|
Cell resting state -65 called |
polarized (negative inside vs outside) - de-polarized/positive = depolarized |
|
|
hyperpolarized |
more negative than resting potential -65 |
|
|
Recording electrode action potential events: |
1. Resting Potential: Cell = -65 2. Threshold: stimulate & give positive charge to -40 3. Rising phase: voltage gated Na channels open & flood cell (rapid depolarization) 4. Overshoot: Na floods in so fast = positive charge of +40 - voltage gated Na channels close - K wants to leave the cell for cell for reach equilibrium (rid of positive charge) - voltage gated K channels open (for 1 ms) 5. Falling phase: 6. Undershoot: cell fully permeable to K = go to equilibrium for K = -80 7. Restore: voltage gated K channel close, Na and K pumps regain equilibrium potential of -65 |
|
|
refractory period |
When action potential cannot occur again - cell must reach negative charge again - can only occur again when K channels close to allow for cell to become negative again |
|
|
Tetradoxin (Fugu Fish) |
Blocks voltage gated Na channels - cell can reach negative state, but Na cannot flood cell and create action potential |
|
|
ACh equation |
Choline + Acetyl Coenzyme A = Acetylcholine + Coenzyme A |
|
|
where ACh equation occurrs & by what |
in mitochondria by Choline Acetyltransferase |
|
|
cycle of ACh |
- Choline + Acetyl Coenzyme A -- ChAT -- = Acetycholine + Coenzyme A in mitochondria - acetylcholine packaged into vesicles via proton antiporters - neuron fires = vesicles fuse with membrane - release ACh into synapse - ACh broke down in the synapse by acetylcholinesterase into original components - choline recycled and reuptake into cell |
|
|
alzheimers due to (neural) |
dificiency in cholinergic system => beta amaloid placques => neurons swell and inflammed |
|
|
possibly alzheimers treatment |
1. Treat cause (reduce placques)-animales
2. Anti inflammatory (no effect) 3. Acetylcholinesterase Inhibitors: enhances time acetylcholine is active in synapse 4. ACh precursor (Choline) 5. Muscarinic Receptor Agonist: bind where ACh should bind = more cholinergic transmission ( 6. Nicotinic Receptor Agonist: more cholinergic transmission |
|
|
Effect of anti inflammatory on Alz |
None |
|
|
Effect of Acetylcholinesterase inhibitors in Alz |
slow cognitive decline up to 6 months Tacrine = non competitive/irreversible & short half life (liver failure) Dorepezil = competitive/reversible & long half life |
|
|
Choline admin |
no effect (if theres enough in the body, the rest eliminated - like Vitamin C>) |
|
|
Effect of Muscarinic receptor agonists |
hard to find specific binding (M1, M2, M3, M4) Melanmaline - peripheral receptors VU100010 = allosteric potentiator, M4 specific |
|
|
Effect of Nicotinic receptor agonist |
not specialized yet, too many peripheral effects |
|
|
How do nerve agents work? |
phosphorylate Acetylcholinesterase in Neuromuscular junto = build up of ACh, not converted and recycled back into choline - excessive ACh bound to Muscarinic receptors = muscles tense = paralyzed |
|
|
Nerve agent antedote |
HI-6 = dephosphorylates AChE Atropine = Muscarinic antagonist (stops ACh binding = releases muscles) |
|
|
ACh Law of Mass action |
Choline + ACoA <=CHaT=> ACh + CoA More ACh being made drives other side of equation |
|
|
3 Principles of ACh Synthesis |
1. Law of mass action 2. End Product inhibition 3. Component availability |
|
|
End product inhibition |
in mitochondria, Choline + ACoA bind to ChAT - the product ACh also binds to ChAT to slow down the catalyst (production) |
|
|
Component availability |
the amount of Choline, ACoA determines how much ACh made |
|
|
How ACh is packaged into vessicles |
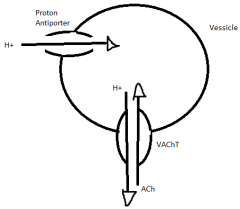
vesicle pumps in H via proton antiporter with ATP; creates concentration gradient; vessicular ACh Transporter (VAChT) uses concentration gradient to kick out H and take in ACh |
|
|
Mechanisms of choline Reuptake |
(LACU) Low affinity Choline uptake - diffusion
(HACU) High affinity Choline uptake - transporter |
|
|
Neuronal aspect of Alzheimers |
degredation of basal forebrain cholinergic neurons projected to neurons in cx & hippocampus |
|
|
Characteristics of Nicotinic receptors |
Excitatory rapid Ionotropic pentameric subunits receptors differ by subunit components binding site usually alpha 10 For NAChR = need two ACh bound to open |
|
|
Characteristics of Muscarinic receptors |
inhibitory & excitatory delay metabotropic 5 receptor types (m1-5) binding site are very similar (differences are not in binding site) Use allosteric binding ligands |
|
|
Subunits of alpha subunit of G protein & what do |
G1 - inhibit G0 - stimulate Gs - other? Gq - PIP2 |
|
|
G-protein cascade pathway |
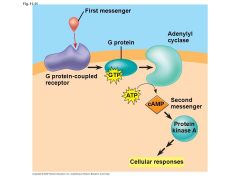
G-protein binds to GDP Alpha subunit detaches & binds to GTP Alpha + GTP bind to Adenylyl cyclase (AC) Use ATP to make cyclic AMP (cAMP) Protein kinase binds to protein Protein is phosphorylated Later stripped of PO4 |
|
|
why myelin? |
less expensive - dont have tons of channels along the axons |