![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
144 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Hepatic encephalopathy clinical presentation |
As an acute confusional state |
|
|
|
Acute confusional state DDX how to differentiate |
All |
|
|
|
Significance of femoral neck fracture on starting bisphosphonate for a postmenopausal female |
ConclusionOur study showed that NSA is effective at predicting the hip fracture risk and that the detection in early post-menopause of a wide NSA together with a low FNBMD should identify females at high probability of incident hip fracture.Hip fracture is the most clinically relevant osteoporotic fracture because it is expensive to treat and has severe consequences [1,2]. Bone mineral density (BMD) measurement at the hip is the strongest predictor of hip fracture [3]. Despite the statistically significant relationship between the femoral neck bone mineral density (FNBMD) and the risk of hip fracture [4], its ability to predict hip fragility fracture does not seem accurate enough for diagnostic purposes [5]. Therefore, hip fragility fracture predictors besides BMD are needed to identify people at risk for fracture prevention [6]. Among these predictors, proximal femur geometry (PFG) parameters have also been proposed, as bone shape adjusts the transmission of the impact forces through the bone, contributing, together with bone structure, to determine the effective stress within the bone [7]. This topic has been largely addressed by using dual energy X-ray absorptiometry (DXA) scans since Beck et al [8] showed the relationship between DXA bone mineral density and femoral neck strength, and Faulkner et al [9] described the association between the hip axis length (HAL) measured by DXA scans and the hip fracture risk. The PFG parameters that have been reported to predict effectively hip fracture independently of BMD are HAL and neck–shaft angle (NSA) [9-18].The majority of these studies are nevertheless cross-sectional [10,12,13], and their results might not have such strong statistical evidence as those derived from longitudinal studies [9,17,18]. In addition, there are some discrepancies among authors about the best PFG parameter to predict the hip fracture risk [19-23]. The aims of this study were therefore to assess and compare in a longitudinal observation the ability of PFG parameters to separate post-menopausal females with hip fracture from those without fracture, and to assess how PFG parameters and BMD are associated with hip fracture incidence, and whether a combination of the two can identify subjects at higher risk of fracture. |
|
|
|
Amlodipine in hypothyroidism induced AF YOU |
Hyperthyroidism, meaning an overactive thyroid, can cause various problems. One of the most serious is atrial fibrillation. This is a form of heart rhythm disturbance in which the upper chambers of the heart, the atria, do not pump effectively. In atrial fibrillation, the heart beats irregularly and often very fast. In this situation the heart does not pump effectively. Symptoms of atrial fibrillation may include fatigue and diminished exercise tolerance, shortness of breath or lightheadedness. The most serious issue is that atrial fibrillation carries a significant risk of stroke.Treating hyperthyroidism with atrial fibrillation involves managing both problems. Depending upon the cause of the hyperthyroidism, a person may require anti-thyroid medication such as methimazole or propylthiouracil, radioactive iodine or surgery. Some forms of hyperthyroidism are temporary and require only symptom management until the condition subsides. Beta blockers–drugs that block adrenaline action -- reduce the symptoms of hyperthyroidism. These medications also lower the heart rate in atrial fibrillation, so that they are useful for both problems.Correction of hyperthyroidism will often lead to resolution of atrial fibrillation. Until that time, or in situations where the atrial fibrillation persists after the hyperthyroidism has been treated, anti-arrhythmic medications may be necessary. These may include beta blockers, as mentioned above, calcium channel blockers such as diltiazem, or various other medications. Patients with atrial fibrillation, whether caused by hyperthyroidism or not, are usually treated with anticoagulants such as warfarin (brand-name Coumadin) as well in order to reduce the risk of stroke.One complicated issue arises when amiodarone is used to treat the atrial fibrillation. This is a powerful anti-arrhythmic medication, often the most effective drug for atrial fibrillation. However, amiodarone contains a large amount of iodine and this may make certain forms of hyperthyroidism worse. Amiodarone may even trigger hyperthyroidism in a person whose atrial fibrillation started for some other reason. Those who suffer from atrial fibrillation and are treated with amiodarone need careful monitoring of their thyroid function. They may need to be seen by endocrinologist as well as a cardiologist to assist with management. |
|
|
|
How effective is bisphosphonates % vise |
Q |
|
|
|
Head tremor in Parkinson disease |
Head tremor is a typical feature of essential tremor. Patients with sporadic Parkinson's disease can have tremor of the tongue, lip, or chin, but classically do not have head tremor. |
|
|
|
Denosumab mechanism of action |
Inhibits RANKL The ligand which activates osteoclasts. |
|
|
|
Significance of BMD on fracture risk by |
.Osteoporosis is a disorder characterized by low bone density and impaired bone strength which is an important risk factor for fracture in older adults. The diagnosis of osteoporosis in postmenopausal women is now based on bone density testing by dual energy x-ray absorptiometry but not other methodologies. However, a specific but arbitrary diagnostic threshold must be distinguished from a strategy to assess fracture risk. In untreated postmenopausal women and older men, bone density is an important, but not the only, determinant for fracture risk. Combining bone density measurements with other independent and validated risk factors for fracture provides a much more accurate assessment of an individual patient's risk for fracture than does bone density alone. The most important of these other risk factors are age and prior fracture history. Clinical guidelines will move away from recommending treatment at specific T scores toward intervention thresholds based on absolute fracture risk. By basing who to treat on fracture probability, therapy can be targeted to those patients who would receive the greatest benefit.
terpretationIn this study, most of the postmenopausal women with osteoporotic fractures had nonosteoporotic bone mineral density values. This finding highlights the importance of considering key clinical risk factors that operate independently of bone mineral density (such as age) when assessing fracture risk.Bone mineral density is commonly used to diagnose osteoporosis and to predict individual fracture risk.1,2 The World Health Organization has proposed a diagnostic classification for bone mineral density based on the T-score (number of standard deviations above or below the mean for young adults), which recognizes 3 categories: normal (T-score –1 or higher), osteopenia (T-score between –1 and –2.5) and osteoporosis (T-score –2.5 or less).3 Although these definitions of osteoporosis and osteopenia were designed as diagnostic thresholds intended for population-based analyses, many clinical guidelines have used them to define thresholds for treatment intervention for individual patients.4,5 Such recommendations may or may not be appropriate, depending on how fracture prevalence varies in relation to the thresholds of bone mineral density.Population-based data on the percentage of fractures that occur in postmenopausal women with either normal bone mineral density or osteopenia are limited. Stone and colleagues,6 in a secondary data analysis of 9704 women over 65 years of age, reported that total hip bone mineral density was more strongly correlated with most fractures than were lumbar spine or peripheral bone mineral density measurements. Moreover, the percentage of fractures among women with osteoporosis (T-score < –2.5) was modest, ranging from less than 10% to 44%.6 Siris and collaborators,7 in a cohort of 149 524 women over 50 years of age, used peripheral bone mineral density to determine the association between osteopenia and self-reported fractures. The prevalence of osteoporosis (T-score –2.5 or less) was 6.4% among the women with fractures, and 18% of all fractures occurred in these women.In the current study, we determined fracture rates and the percentage of fractures among postmenopausal women in relation to bone mineral density at different central sites and in relation to the cut points defined by the World Health Organization; we also determined how fracture patterns differed between women 50–64 years of age and those 65 years of age and older ConclusionOur study showed that NSA is effective at predicting the hip fracture risk and that the detection in early post-menopause of a wide NSA together with a low FNBMD should identify females at high probability of incident hip fracture.Hip fracture is the most clinically relevant osteoporotic fracture because it is expensive to treat and has severe consequences [1,2]. Bone mineral density (BMD) measurement at the hip is the strongest predictor of hip fracture [3]. Despite the statistically significant relationship between the femoral neck bone mineral density (FNBMD) and the risk of hip fracture [4], its ability to predict hip fragility fracture does not seem accurate enough for diagnostic purposes [5]. Therefore, hip fragility fracture predictors besides BMD are needed to identify people at risk for fracture prevention [6]. Among these predictors, proximal femur geometry (PFG) parameters have also been proposed, as bone shape adjusts the transmission of the impact forces through the bone, contributing, together with bone structure, to determine the effective stress within the bone [7]. This topic has been largely addressed by using dual energy X-ray absorptiometry (DXA) scans since Beck et al [8] showed the relationship between DXA bone mineral density and femoral neck strength, and Faulkner et al [9] described the association between the hip axis length (HAL) measured by DXA scans and the hip fracture risk. The PFG parameters that have been reported to predict effectively hip fracture independently of BMD are HAL and neck–shaft angle (NSA) [9-18].The majority of these studies are nevertheless cross-sectional [10,12,13], and their results might not have such strong statistical evidence as those derived from longitudinal studies [9,17,18]. In addition, there are some discrepancies among authors about the best PFG parameter to predict the hip fracture risk [19-23]. The aims of this study were therefore to assess and compare in a longitudinal observation the ability of PFG parameters to separate post-menopausal females with hip fracture from those without fracture, and to assess how PFG parameters and BMD are associated with hip fracture incidence, and whether a combination of the two can identify subjects at higher risk of fracture. |
|
|
|
Osteonecrosis of the jaw what causes it oral# bisphosphonates |
Q |
|
|
|
Q Recognized fx of PMRWeakness of distal muscle groupsElevation of CPKAn association with bronchial CAWt lossPeak incidence in the fourth decade of life
|
ResultsIn recent years, few studies have been published regarding the relationship between PMR and cancer. In 2010, Ji et al5 examined the overall and specific cancer risks among Swedish subjects following hospitalization for PMR and giant cell arteritis (GCA) and noted that the risk of cancer was highest in the first year after hospitalization. In particular, of 3941 total cancer diagnoses, 783 (19.1%) were in the first year. Research Database (GPRD) highlighted that elderly patients with a PMR diagnosis were significantly more likely to receive a cancer diagnosis in the year after PMR diagnosis Diagnosis of PMR was supported by the requirement that patients had received at least two prescriptions for corticosteroids following diagnosis; however, no data regarding response to treatment were available.6 In 2014, Ungprasert et al7 published a systematic review including six studies with pooled statistical analysis revealing a 14% excess risk of malignancy in patients with GCA/PMR. The risk of malignancy appeared to be higher in the first 6–12 months after diagnosis. However, when sensitivity analysis was performed excluding one of the six studies due to potential selection bias, the pooled risk ratio decreased to 8% and did not achieve statistical significance.7 In a series of 200 PMR patients consecutively observed in our geriatric rheumatologic outpatient clinic from 2004 to 2014, we have observed 51 cancer cases (Table 1).Table 1Table 1Cancer in our cohort of PMR patients.The majority of malignancy was diagnosed after >5 years from initial diagnosis of PMR. The incidence rate was not different from that of a homogeneous non-PMR population. Only five of these (prostate cancer, vesical cancer, multiple myeloma, gastric neuroendocrine gastrin-secreting tumor, and adenocarcinoma of the lung, highlighted in bold in Table 1) were observed in the first year after diagnosis of PMR with a percentage equal to 9.8%. Three of these had a remitting seronegative symmetrical synovitis with pitting edema (RS3PE syndrome) as part of the PMR clinical picture.8 Our series does not include the elderly with GCA. GCA has per se a neoplastic risk. The very low percentage of cancer diagnosed in our cohort during the first year after the diagnosis of PMR compared to the much higher percentages observed in the two cited studies (9.8% vs 19.1% and vs 69%, respectively) highlights the diagnostic set |
|
|
|
Q Predictors of possible fatal outcome in pneumonia in the elderlyMultilobar involvementStreptococcus pneumonia causing pneumoniaLow serum sodium levelsNew development of AFBacteraemia
|
Q |
|
|
|
Q Following statements are true regard to renal failure in M Myeloma
It is seen in about 25 – 35% of cases
About 5% will have severe renal failure
Renal failure is more commonly caused by hypercalcaemia
Bence-Jones proteinuria causing renal tubular damage is the commonest cause for renal failure
Other factors which could contribute to renal failure are NSAID therapy & dehydration
|
Renal failure is a frequent complication in patients with multiple myeloma (MM) that causes significant morbidity.
In the majority of cases, renal impairment is caused by the accumulation and precipitation of light chains, which form casts in the distal tubules, resulting in renal obstruction.
In addition, myeloma light chains are also directly toxic on proximal renal tubules, further adding to renal dysfunction.
Adequate hydration, correction of hypercalcemia and hyperuricemia and antimyeloma therapy should be initiated promptly.
Recovery of renal function has been reported in a significant proportion of patients treated with conventional chemotherapy, especially when high-dose dexamethasone is also used.
Severe renal impairment and large amount of proteinuria are associated with a lower probability of renal recovery.
Novel agents, such as thalidomide, bortezomib and lenalidomide, have significant activity in pretreated and untreated MM patients. Although there is limited experience with thalidomide and lenalidomide in patients with renal failure, data suggest that bortezomib may be beneficial in this population. Clinical studies that have included newly diagnosed and refractory patients indicate that bortezomib-based regimens may result in rapid reversal of renal failure in up to 50% of patients and that full doses of bortezomib can be administered
Renal impairment (RI) is a common complication of multiple myeloma (MM).
Around 50% of patients with MM have RI at presentation, and up to 5% require dialysis treatment.
Severe acute kidney injury (AKI) as a cause of RI is a particular challenge as historically the survival of patients who sustain this complication and require dialysis is very poor. However, in this current period, survival is improving and the focus is on optimum use of novel chemotherapies and the evaluation of extra-corporeal therapies for removal of serum immunoglobulin light chains. |
RI in patients with MM is commonly associated with excess monoclonal free light chain (FLC) production; myeloma cast nephropathy is the predominant renal pathology in patients presenting with severe RI secondary to AKI.
The majority of patients have mild to moderate RI and recover renal function.
However, patients with more severe RI, in particular those with a requirement for dialysis, are less likely to recover renal function.
Rapid diagnosis and prompt institution of anti-myeloma therapy is an important determinant of renal function recovery, through targeting early and sustained reduction of involved monoclonal FLC.
Novel agents are associated with excellent disease response, and bortezomib is now widely used as a first-line agent in the management of MM in patients with severe RI. Extended haemodialysis using high cut-off dialysers is more effective for extracorporeal removal of FLC than plasma exchange, and clinical trials are in process. High-dose chemotherapy with autologous stem cell transplantation does have a role in patients with severe RI but requires careful patient selection. Key Messages RI is very common in patients with MM, and renal function recovery is associated with improved clinical outcomes. The commonest cause of severe RI in patients with MM is myeloma cast nephropathy. (2) The efficacy of novel treatments (bortezomib, carfilzomib, thalidomide, and lenalidomide) has predominantly been assessed in Western patients. Bortezomib and dexamethasone are the current standard of care for MM and severe RI in the West. Severe RI is not a contraindication to autologous stem cell transplantation (ASCT). Most of the data are from the West; there are case reports from China describing good outcomes with ASCT. The removal of FLC by high-cut-off hemodialysis is under evaluation in randomized controlled trials (RCTs) in the West. Fifty percent of patients have renal impairment (RI) at presentation, and up to 20% have severe acute kidney injury (AKI). Severe AKI is usually a consequence of myeloma cast nephropathy (MCN), caused by high levels of immunoglobulin free light chain (FLC). The presence of severe AKI is an important complication of MM as it is associated with an increased risk of early mortality; however, the impact of mild-to-moderate RI at presentation on patient outcome is unclear. Recent advances in chemotherapy have led to better overall survival (OS) and for patients who require dialysis as a consequence of MM, increased recovery rates of independent renal function are being reported, and these improved renal outcomes are associated with better OS.or end-stage kidney disease, the area under the concentration-time curve increases by approximately 185–420%, and the median half-life increases by 6–12 h; furthermore, a single session of 4-hour haemodialysis only removes 31% of the administered dose [96]. As a result, dose recommendations in patients with MM and RI are in place [97]. Lenalidomide therapy is associated with an increased risk of thromboembolic events [91], and neutropenia and thrombocytopenia are also important side effects [98].Lenalidomide and dexamethasone were assessed in patients with RI in the MM-009 and MM-010 multicentre, phase III clinical trial; 71, 24 and 5% had a CrCl of ≥60, 30–59 and <30 ml/min, respectively. There was no significant difference in the overall response, time to progression and progression-free survival between groups. However, patients with a CrCl <30 ml/min had a shorter survival (18.4 months) than those with a CrCl 30–59 ml/min (29.0 months) or CrCl ≥60 ml/min (38.9 months). A renal response occurred in 72%. Thrombocytopenia and discontinuation of lenalidomide (mainly due to cytopenia) was more common in those with severe RI [99].de la Rubia et al. [100] reported the use of lenalidomide and dexamethasone in 15 refractory and/relapsed MM patients on dialysis. Thirteen patients received lenalidomide three times a week after dialysis treatment at a dose of 15 mg/day, and 2 patients received lenalidomide at a dose of 5 mg/day and 5 mg on alternate days, respectively. 29% achieved a CR, 7% a VGPR and 29% a PR. One patient who achieved a PR attained independence from dialysis. Haematological toxicity was common; 53% of patients required a reduction in the dose of lenalidomide due to the development of cytopenia. This study showed that with careful monitoring and with appropriate dose adjustments, lenalidomide could be used for relapsing or refractory MM in patients requiring dialysis. Overall, the results of studies to date indicate that lenalidomide therapy is an important treatment option in the management of MM in patients with RI [101,102,103,104,105].Pomalidomide Pomalidomide is another new-generation immunomodulatory agent used in combination with dexamethasone for the management of relapsed and/or refractory MM. The superiority of pomalidomide with low-dose dexamethasone compared to pomalidomide alone or high-dose dexamethasone alone was reported in a phase II [106] and a phase III [107] study, respectively; however, individuals with moderate to severe RI at presentation were excluded from both studies. It is metabolised extensively via multiple pathways, with <5% of the administered pomalidomide dose excreted unchanged in the urine. Studies to assess the efficacy and safety of pomalidomide in patients with severe RI are currently underway [108].Autologous Stem Cell TransplantationHigh-dose chemotherapy followed by ASCT is an established treatment option for MM. RI had no impact on stem cell collection and post-transplant engraftment in small single-centre studies [109,110], with some centres reporting cases of recovery from requiring dialysis [110,111].High-dose melphalan (200 mg/m2) is associated with excessive toxicity; when the dose of melphalan was reduced to 140 mg/m2 in a study where 21 of the 81 patients had advanced RI (creatinine >177 μmol/l with 47% on dialysis), transplant-related mortality was observed in 6 and 13% of patients after single and tandem ASCT, respectively. Dialysis dependence and melphalan dose did not affect event-free survival or OS [112]. In a subsequent study in dialysis patients, a further reduction in melphalan dose (100 mg/m2) produced a similar toxicity profile, transplant-related mortality, disease response and OS compared to patients with no RI [113].Published data around the disease and renal response in patients with advanced RI are variable. In a single-centre study of 59 patients requiring dialysis, ASCT was associated with independence of dialysis in 13 of 54 patients who survived more than 30 days. A shorter duration of dialysis (≤6 months), achievement of a CR or near CR and CrCl ≥10 ml/min were all associated with recovery of independent kidney function [114.]More recently, Mayo clinic investigators reported long-term outcomes following ASCT in 30 patients with advanced RI (creatinine >3 mg/dl); 15 patients were receiving dialysis, and only 1 recovered renal function. The non-dialysis patients had a modest improvement in eGFR from 15 to 19 ml/min. CR was noted in 14 patients. Although patients who achieved a CR had a better median eGFR than those who did not, the authors found no association between haematological response and baseline eGFR with renal outcome [115].Current guidelines support the idea that ASCT is an attractive treatment option in the management of MM; however, careful patient selection is required, especially in patients with advanced RI [116].Role of Extra-Corporeal Removal of FLCTherapeutic Plasma Exchange The largest randomised control trial in patients with MM and severe AKI recruited between 1998 and 2003 failed to show a beneficial effect of plasma exchange (PE) on patient and renal outcomes; the limitations of this study included the lack of a renal biopsy confirming a diagnosis of MCN and the absence of serum or urinary light chain measurement [117].Subsequently, Leung et al. [118] investigated the efficacy of PE in 40 patients with MM and severe RI that included 9 (22.5%) patients on dialysis. A renal response was noted in 18 (45%) patients, which included 14 patients with MCN who had sFLC levels measured before and after treatment with PE. Of these 14 patients, an sFLC reduction of ≥50% was reported in 64.3%; 7 of these patients had a significant renal response. Two (22%) patients subsequently became dialysis independent. The authors concluded that PE for extra-corporeal FLC removal only had a role in biopsy-proven MCN and in patients who demonstrated a ≥50% reduction in sFLC levels from baseline.However, PE as an isolated treatment does not provide clinical benefit, and the improvements in clinical outcome associated with a ≥50% reduction in FLC in this study most likely reflect a chemosensitive FLC clone. As FLC have a molecular weight of 25–50 kDa and are distributed in both the intra- and extravascular compartments, a single session of PE removes <10% of extracellular FLC. |
|
|
Q Following statements are true regard to renal failure in M MyelomaIt is seen in about 25 – 35% of casesAbout 5% will have severe renal failureRenal failure is more commonly caused by hypercalcaemiaBence-Jones proteinuria causing renal tubular damage is the commonest cause for renal failureOther factors which could contribute to renal failure are NSAID therapy & dehydration
|
Q |
|
|
|
Q 31.A man of 56yrs who drinks one pint of beer daily has a history of recurrent renal stones. He also suffered painful swelling of right foot about one year ago. His GP had put him on hydrochlorothiazide when his BP was found high.What practical points would agree with his managementHe is likely to have hyperuricaemia and will need allopurinolBeer drinking is acceptable as it falls within safe limitHCT is an ideal antihypertensiveHis renal functions should be assessed before starting uricosuric drugs
|
Q |
|
|
|
PMR age of onset |
Over 50$ |
|
|
|
A 65 yr old vendor got admitted to the hospital with a hx of sudden loss of consciousness lasting apparently for 2min.An eye witness account had been him turning pale without getting fits. There has been no incontinence of urine. He admitted having a similar episode four months ago. He is a smoker and denied having ny significant past hx. His BP 130/76 and systemic examination was normal. Likely possibilities Stokes Adams attacksEpilepsy Vasovagal attackBrain stem ischaemia Hypoglycaemia
|
Q |
|
|
|
A lady of 65yrs complained of feeling unwell with loss of appetite. She also had pain in her shoulder & intermittent headache. Which of following would adequately discuss her caseShe would have difficulty in combing her hairTenderness over temporal area expectedBlood investigations would reveal a microcytic anaemiaTemporal artery biopsy is always indicatedShe can suddenly go blind
|
Q |
|
|
|
65yr old lady was admitted in a state of coma to the PCU of a District General Hospital. On examination she was pale & puffy with dry skin recorded temperature of 35.6 celcius .GCS 7/17 pulse 40 bpmThe most likely diagnosis would be drug overdose An important step in management would be to cover her immediately with warm balnkets and warm herHypoglycaemia should be exclude at once Hypernatraemia is a known association L Thyroxine should be started through NG tube at smaller doses
|
Management Investigations Causes
Medical CareMyxedema coma is a medical emergency that requires immediate attention. If the diagnosis is suspected, immediate management is necessary before confirming the diagnosis due to the high associated mortality rate. Patients with myxedema coma should be managed in an intensive care unit with continuous cardiac monitoring. Initial steps in management include the elements below. Airway management Maintenance of adequate airway is crucial, since most patients have depressed mental status along with respiratory failure.
Mechanical ventilation is commonly required during the first 36-48 hours, but some patients require prolonged respiratory support for as long as 2-3 weeks.
Thyroid hormone replacementThe ideal mode of therapy and doses of thyroid hormone therapy in myxedema coma remain controversial due to the rarity of the condition and lack of clinical trials. Some clinicians favor the administration of levothyroxine (T4), while others prefer a combination of T4 and liothyronine (T3). [1, 2, 28, 29] The American Thyroid Association recommends combination therapy with T4 and T3. [30]Because of reduced gastrointestinal absorption, intravenous thyroid hormone therapy is advised.An intravenous loading dose of 300-600 micrograms of levothyroxine (T4) is followed by a daily intravenous dose of 50-100 micrograms. [2] Larger doses of T4 probably have no advantage and may be dangerous. [31] The lower end of the dosing range is recommended in older patients, those at risk for cardiac complications such as myocardial infarction and arrhythmias, and in patients with coronary artery disease, since full-dose T4 therapy may worsen myocardial ischemia by increasing myocardial oxygen consumption. [30]Because the rate of conversion of T4 to the active hormone T3 can be reduced in these patients, the addition of T3 along with T4 has been recommended. [30] T3 has a quicker onset of action than T4, as increases in body temperature and oxygen consumption has been reported to be faster with T3 therapy compared to T4. [2] T3 therapy is given as bolus of 5-20 micrograms intravenously and to be continued at a dosage of 2.5-10 micrograms every 8 hours depending on the patient's age and coexistent cardiac risk factors. [30]Intravenous levothyroxine treatment in severely hypothyroid patients usually leads to improvement in cardiovascular, renal, pulmonary, and metabolic parameters within a week. Serum T4 and T3 concentrations may improve or normalize with a similar time frame, with more gradual improvement in serum TSH. Thus, the therapeutic endpoints in myxedema coma should be improved mental status, improved cardiac function, and improved pulmonary function.
Measurement of thyroid hormones every 1-2 days is suggested. [30] Failure of TSH to decrease or of thyroid hormone levels to increase suggests the need to increase doses of T4 and/or add T3.The treatment is changed to the oral form once the patient is able to take medications by mouth.
Glucocorticoid therapy Patients with primary hypothyroidism may have concomitant primary adrenal insufficiency while patients with secondary hypothyroidism may have associated hypopituitarism and secondary adrenal insufficiency. The other rationale for the treatment with corticosteroids is the potential risk of precipitating acute adrenal insufficiency caused by the accelerated metabolism of cortisol that follows T4 therapy. [2]Stress doses of intravenous glucocorticoids should be administered until the possibility of adrenal insufficiency is excluded by a random serum cortisol, which is helpful only if very low, or, better, by an ACTH stimulation test.Hydrocortisone at a dose of 50-100 mg every 8 hours is administered. An alternative is dexamethasone at a dose of 2-4 mg every 12 hours. Dexamethasone has the advantage of not affecting the serum cortisol concentration and can be used immediately without affecting the results of the ACTH stimulation test, which can be performed at any time. If the test is normal, corticosteroids can be stopped without tapering.A study by Ren et al indicated that pretibial myxedema can be effectively treated with multipoint intralesional injections of compound betamethasone. The investigators found that after one treatment, 21.7% of patients achieved complete remission, while two, three, and four treatments were followed by complete remission in 34.8%, 17.4%, and 17.4% of patients, respectively. [32]
Supportive measures Treat hypothermia with passive rewarming using ordinary blankets and a warm room. Active rewarming using external devices carries a risk of vasodilatation and worsening hypotension and should be avoided. The use of a rectal probe helps to determine the true core temperature and to monitor rewarming.
Treat associated infection. Given the severity of the condition, infection should always be considered and empiric broad-spectrum of antibiotics be considered until appropriate cultures are proven negative.
Correct severe hyponatremia with saline and free water restriction.
Correct hypoglycemia with intravenous dextrose.
Hypotension is usually corrected with thyroid hormone therapy. If blood pressure continues to be low, cautious use of intravenous fluids with normal saline is advised.
Refractory hypotension can be treated with vasopressors such as dopamine, but patients should be weaned off the vasopressor as soon as possible because of the risk of pressor-induced ischemic event. Patients who are awake, no longer dependent on a ventilator, and medically stable may be transferred from the intensive care unit to a medical ward.
Surgical Care Patients with myxedema coma who require surgical intervention are considered high risk for complications of anesthesia as well as intraoperative and postoperative complications. Stabilization of these patients before proceeding to surgery is preferred unless the procedure is urgent.
In life-threatening situations, the loading dose of T4 and glucocorticoids are administered before induction of anesthesia.
Careful administration of anesthetic agents with consideration of using lower doses should be exercised given the decreased metabolism of these agents in patients with myxedema coma
.Close monitoring during surgery and in the postoperative period in a critical care unit is imperative. Monitoring includes respiratory, cardiac, and volume and temperature status
Consultations Consultations include endocrinologists and critical care specialists. Depending on complications, consultations with pulmonologists and/or cardiologists may be appropriate.
Diet Most patients will be initially ill and will not be given any food by mouth. Many patients require nasogastric feeding, and if mechanical ventilation is prolonged, total parenteral nutrition may be required.
Activity Once stable, patients may progress to usual activity as their strength allows. Physical therapy may be needed for incapacitated patients.
Prevention Patients with a history of thyroid resection or ablation for hyperthyroidism and persons with a history of Hashimoto thyroiditis are at risk for developing hypothyroidism, and the TSH level should be monitored yearly. Such patients should be informed that hypothyroidism could occur in the future. They should understand the symptoms that signal the condition and the need to seek medical attention for appropriate testing. |
In cold climates, inadequately heated residences are a significant cause of myxedema coma/crises in patients with undiagnosed or inadequately treated hypothyroidism. Thyroid function tests should be monitored regularly in patients with hypothyroidism until the appropriate dose of levothyroxine is reached. Adherence to thyroid hormone therapy should be assessed regularly and to ensure maintenance of euthyroid state. Patients who are deemed nonadherent or have issues that may hinder adherence should have their thyroid function closely monitored. Patients are advised to report to their physicians if they are prescribed any new medications since some drugs may interfere with the absorption, production, secretion, or clearance of thyroid hormone therapy. Patients should also contact their health care provider if symptoms of inadequately-treated hypothyroidism persist. Long-Term Monitoring Follow-up care after discharge is necessary to ensure adherence with thyroid hormone replacement .If primary hypothyroidism was diagnosed, TSH levels are assessed every 4-6 weeks, and the dose of T4 is adjusted accordingly. If hypothyroidism is secondary to pituitary dysfunction, free T4 levels are monitored. TSH level is not an accurate measure of thyroid function in this setting. |
|
|
Are there contraindications for primary PCI in MI |
Contraindications Special considerations While primary PCI is extremely valuable in most patients with STEMI, certain special scenarios may exist and dictate an alternative strategy.
Some patients are not good candidates for reperfusion therapy in general. Old, debilitated patients with advanced dementia, few symptoms, and hemodynamically insignificant myocardial infarction (MI) may be best treated with appropriate medical therapy.
Similarly, those with MI that occurred more than 24 to 36 hours prior to arrival, and who are in extremis from a hemodynamic point of view may be beyond salvage, particularly if they are very old and debilitated.
Interventional procedures in these cases may only hasten the inevitable.
Patients with witnessed cardiac arrest and resuscitation in the field should be taken immediately for primary PCI, if STEMI is present, irrespective of neurological status. Consideration should be given, though, to utilization of cooling protocols to improve chances for neurological recovery. Occasionally, the interventional team cannot be assembled in a timely fashion because of ongoing high-risk cases or circumstances beyond control (extreme weather, traffic, etc.) Then, patients should be quickly evaluated for fibrinolysis and treated with it, if suitable candidates. Definitive mechanical therapy can be provided at a later time, without compromising the potential benefit of timely reperfusion. |
|
|
|
Causes of elevated troponin levels |
M |
|
|
|
Morphine |
Morphine relieves breathlessness due to CHF. A larger study is indicated Morphine 2 to 4 mg IV, repeated q 15 min as needed, is highly effective but can depress respiration, can reduce myocardial contractility, and is a potent venous vasodilator.
Evidence also suggests that morphine use interferes with some P2Y12 receptor inhibitors. A large retrospective trial showed that morphinemay increase mortality in patients with acute myocardial infarction Hypotension and bradycardia secondary to morphine can usually be overcome by prompt elevation of the lower extremities. |
|
|
|
It's true |
Fibrinolytics are not indicated for any NSTEMI patients. Risk outweighs potential benefit. |
|
|
|
What is measuring routinely and periodically mean |
Q |
|
|
|
Warfarin |
INR range and treatment duration• Maintain an INR of 2.0-3.0• Surgery-provoked DVT or PE: Treatment duration of 3 months •Transient (reversible) risk factor-induced DVT or PE: Treatment duration of 3 months •First unprovoked proximal DVT or PE with low or moderate bleeding risk: Extended treatment consideration with periodic (ie, annual) risk-benefit analysis •First unprovoked proximal DVT or PE with high bleeding risk: Treatment duration of 3 months •First unprovoked distal DVT regardless of bleeding risk: Treatment duration of 3 months •Second unprovoked DVT or PE with low or moderate bleeding risk: Extended treatment •Second unprovoked DVT or PE with high bleeding risk: Treatment duration of 3 months •DVT/PE and active cancer: Extended treatment, with periodic risk-benefit analysis (ACCP recommends LMWH over vitamin K antagonist therapy) •Prevention of venous thromboembolism for total knee arthroplasty, total hip arthroplasty, and hip fracture surgery: Minimum treatment duration of 10-14 days, with a recommendation to extend outpatient therapy to 35 days (ACCP recommends LMWH over vitamin K antagonist therapy)Stroke &
|
|
|
|
Warfarin |
Indications for indefinite treatment duration •Persistent or paroxysmal nonvalvular AF in patients with a high risk of stroke: Ie, patients who have risk factors for stroke, such as prior ischemic stroke, transient ischemic attack, or systemic embolism or who have 2 of the following risk factors--age greater than 75 years, moderately or severely impaired left ventricular systolic function and/or heart failure, history of hypertension, or diabetes mellitus •Persistent or paroxysmal nonvalvular AF in patients with an intermediate risk of ischemic stroke: Ie, patients who have 1 of the following risk factors--age >75 years, moderately or severely impaired left ventricular systolic function and/or heart failure, history of hypertension, or diabetes mellitus •AF and mitral stenosis •≥2 episodes of documented DVT or PE
|
|
|
|
New guidelines on cholesterol management |
🏀 In adults 40 to 75 years of age without diabetes mellitus and with LDL-C levels ≥70 mg/dL (≥1.8 mmol/L) at a 10-year ASCVD risk of ≥7.5%, start a moderate-intensity statin if a discussion of treatment options favors statin therapy. Risk-enhancing factors favor statin therapy (see No. 8). If risk status is uncertain, consider using coronary artery calcium (CAC) to improve specificity (see No. 9). If statins are indicated, reduce LDL-C levels by ≥30%, and if 10-year risk is ≥20%, reduce LDL-C levels by ≥50%. 🏀 In adults 40 to 75 years of age without diabetes mellitus and 10-year risk of 5% to 19.9%, riskenhancing factors favor initiation of statin therapy (see No. 7). Risk-enhancing factors include family history of premature ASCVD; persistently elevated LDL-C levels ≥160 mg/dL (≥4.1 mmol/L); metabolic syndrome; chronic kidney disease; history of preeclampsia or premature menopause (age <40 years); chronic inflammatory disorders (eg, rheumatoid arthritis, psoriasis, or chronic HIV); high-risk ethnic groups (eg, South Asian); persistent elevations of triglycerides ≥175 mg/dL (≥1.97 mmol/L); and, if measured in selected individuals, apolipoprotein B ≥130 mg/dL (≥2500 nmol/L), high-sensitivity C-reactive protein 2.0 mg/L (190 nmol/L), ankle brachial index <0.9, and lipoprotein (a) ≥50 mg/dL (125 nmol/L), especially at higher values of lipoprotein (a). 🏀 In adults 40 to 75 years of age without diabetes mellitus and with LDL-C levels ≥70 to 189 mg/dL (≥1.8 to 4.9 mmol/L), at a 10-year ASCVD risk of ≥7.5% to 19.9%, if a decision about statin therapy is uncertain, consider measuring CAC. If CAC is zero, treatment with statin therapy may be withheld or delayed, except in cigarette smokers, those with diabetes mellitus, and those with a strong family history of premature ASCVD. A CAC score of 1 to 99 favors statin therapy, especially in those >55 years of age. For any patient, if the CAC score is ≥100 Agatston units or ≥75th percentile, statin therapy is indicated unless otherwise deferred by the outcome of clinician-patient risk discussion. 🏀Assess adherence and percentage response to LDL-C-lowering medications and lifestyle changes with repeat lipid measurement 4 to 12 weeks after statin initiation or dose adjustment, repeated every 3 to 12 months as needed. Define responses to lifestyle and statin therapy by percentage reductions in LDL-C levels compared with baseline. In ASCVD patients at very high risk, triggers for adding nonstatin drugs are defined by threshold LDL-C levels ≥70 mg/dL (≥1.8 mmol/L) on maximal statin therapy (see No. 3). |
|
|
|
Is Chondrocalcinosis is invariably present in pseudogout q |
View Outline ToolsClinical manifestations and diagnosis of calcium pyrophosphate crystal deposition (CPPD) diseaseThe content on the UpToDate website is not intended nor recommended as a substitute for medical advice, diagnosis, or treatment. Always seek the advice of your own physician or other qualified health care professional regarding any medical questions or conditions. The use of UpToDate content is governed by the UpToDate Terms of Use. ©2019 UpToDate, Inc. All rights reserved.Author:Ann K. Rosenthal, MD, FACPSection Editor:Nicola Dalbeth, MBChB, MD, FRACPDeputy Editor:Paul L Romain, MDContributor DisclosuresAll topics are updated as new evidence becomes available and our peer review process is complete.Literature review current through: Feb 2019. | This topic last updated: Jul 24, 2018.INTRODUCTION — Precipitation of crystals of calcium pyrophosphate dihydrate (CPP) in connective tissues may be associated with several clinical syndromes, but is sometimes asymptomatic. The consequences of CPP deposition include acute inflammatory arthritis, inflammatory and degenerative chronic arthropathies, and radiographic cartilage calcification and constitute the spectrum of calcium pyrophosphate crystal deposition (CPPD) disease [1-3].The clinical manifestations and diagnosis of CPPD disease are discussed here. The pathogenesis and etiology of this disorder and the treatment of CPPD diseases are discussed separately. (See "Pathogenesis and etiology of calcium pyrophosphate crystal deposition (CPPD) disease" and "Treatment of calcium pyrophosphate crystal deposition (CPPD) disease".)TERMINOLOGY — The names traditionally used for calcium pyrophosphate dihydrate (CPP) crystal deposition (CPPD) diseases include pseudogout, chondrocalcinosis, and pyrophosphate arthropathy. Based upon a review of the relevant literature, however, a European League Against Rheumatism (EULAR) consensus panel suggested alternative terminology (see 'Clinical manifestations' below); the panel also reviewed diagnostic approaches to these conditions and the evidence supporting these approaches [4].In view of its wider acceptance in the literature since the introduction of this terminology in 2011, we will place primary emphasis here on the EULAR task force terminology [4], in which the term "calcium pyrophosphate crystal deposition" (abbreviated as "CPPD") is proposed as the umbrella term for all instances of calcium pyrophosphate crystal occurrence, ie, the presence of crystals. Symptoms may or may not be present in patients with CPPD, and the term CPPD disease suggests the presence of arthritis.Nevertheless, despite their limitations, the clinical syndromes and findings implied by the traditional terms for CPPD disease are likely to be retained to a greater or lesser extent by some clinicians [5,6]. Familiarity with the older nomenclature may, in addition, be useful in instances where literature searches using only EULAR task force terms overlook citations indexed under the traditional terms. These include:●Pseudogout – Pseudogout accurately describes acute attacks of CPPD-induced synovitis, which clinically resemble acute attacks of urate gout. However, the majority of individuals with CPPD never experience such episodes, and the range of clinical events characterizing gout and CPPD disease extend well beyond those that characterize acute gouty arthritis. For these reasons, the EULAR consensus panel prefers the term "acute calcium pyrophosphate (CPP) crystal arthritis" instead of pseudogout.Notably, however, the use of the term "pseudogout" has also been used more broadly to direct attention to the shared clinical features of the two major crystal-induced arthritides (gout and pseudogout) that are seen much less commonly in other joint diseases, such as rheumatoid arthritis (RA) or spondyloarthritis. These features include recurrent, usually monoarticular, flares of inflammatory arthritis that are self-limited in duration and most often resolve completely. Retention of the diagnostically useful concept of a shared clinical profile of crystal-induced arthritis has merit, as does the mandate to distinguish gout from pseudogout with respect to therapeutic approaches. Thus, use of the term "pseudogout" for acute CPP crystal arthritis will likely persist.●Chondrocalcinosis – Chondrocalcinosis refers to radiographic calcification in hyaline and/or fibrocartilage (image 1). It is commonly present in patients with CPP crystal deposition disease but is neither absolutely specific for CPPD nor universal among affected patients. The EULAR panel designates this finding as "cartilage calcification (CC)."EPIDEMIOLOGY — Calcium pyrophosphate crystal deposition (CPPD) has been estimated to affect 4 to 7 percent of the adult populations of Europe and the United States [7,8], especially among persons of advanced age. However, an estimate of the prevalence of clinically significant CPPD disease has been more difficult to attain, in large part because the available prevalence estimates have relied primarily upon radiographically detected cartilage calcification rather than clinical evaluation, and also because prevalence data concerning patients less than about 60 years of age are not available.The average age at diagnosis of CPPD disease in one study was 72 years [3]. Radiographic surveys have demonstrated an age-related increase in the prevalence of cartilage calcification [9,10]. This was illustrated in a report that supplemented radiographs of the knees with radiographs of the hands, wrists, and pelvis [10]. The prevalence of radiographic calcium pyrophosphate deposition according to age, among 100 consecutive patients admitted to an acute geriatric unit, was:●65 to 74 years – 15 percent●75 to 84 years – 36 percent●>84 years – Almost 50 percentThe gender distribution of CPPD disease has differed among large series [3,11,12], but no major gender predominance appears likely. Attacks of acute arthritis may occur more frequently in men, while typical osteoarthritis (OA) with calcium pyrophosphate crystal deposition or the atypical pattern of OA characteristic of CPPD are more common in women.CLINICAL MANIFESTATIONS — The majority of individuals with calcium pyrophosphate (CPP) crystal deposition (CPPD) are asymptomatic with respect to joint involvement, and there is considerable diversity in the patterns of joint disease among those who develop symptoms. A clinical classification of CPPD disease emerged from the studies of McCarty [1] and colleagues [2,3], which drew particular attention to the capacity for the clinical manifestations of this disorder to mimic virtually any type of arthritis, including gout, rheumatoid arthritis (RA), osteoarthritis (OA), and neuropathic joint disease. This clinical classification (in parentheses below) relates to the nomenclature proposed by the European League Against Rheumatism (EULAR) task force [4] as follows:●Asymptomatic CPPD disease ("asymptomatic CPPD") (see 'Asymptomatic CPPD disease' below)●Acute CPP crystal arthritis ("pseudogout") (see 'Acute CPP crystal arthritis' below)●Chronic CPP crystal inflammatory arthritis ("pseudo-RA") (see 'Chronic CPP crystal inflammatory arthritis' below)●OA with CPPD, with or without superimposed acute attacks ("pseudo-OA") (see 'Osteoarthritis with CPPD' below)●Severe joint degeneration (pseudo-neuropathic joint disease) (see 'Severe joint degeneration' below)●Spinal involvementAsymptomatic CPPD disease — Most joints in which CPP crystal deposition is readily apparent on radiographs are asymptomatic, even among patients in whom acute or chronic clinical manifestations of CPPD disease in one or several other joints have occurred. However, patients with apparent asymptomatic CPPD may be found to have manifestations of an arthritic disorder upon close questioning. As an example, in one series of older patients with radiographic but ostensibly asymptomatic CPP crystal deposition, a higher frequency of wrist complaints and genu varus deformity was reported by questionnaire and detected by examination, respectively, than in a control group of similar age but without radiographic chondrocalcinosis [9].Acute CPP crystal arthritis — "Acute CPP crystal arthritis" is characterized by self-limited acute or subacute attacks of arthritis involving only one or several extremity joints [12]. The traditional term pseudogout underlines the usually close resemblance of these attacks to those of urate gout, in the accompanying symptoms and signs of severe acute inflammation (intense pain, redness, warmth, swelling, and joint disability), and in the occasional occurrence of synchronous inflammation of several adjacent joints (cluster attacks) or, conversely, petite attacks (which are minimally painful episodes of joint warmth and swelling).The knee is affected in over 50 percent of all acute attacks of acute CPP crystal arthritis, while the first metatarsophalangeal (MTP) joint is the most frequently affected in urate gout. Other joints typically affected in acute CPP crystal arthritis include wrists, shoulders, ankles, feet, and elbows. Initial episodes of acute CPP crystal arthritis may persist longer before remitting than the one or two weeks commonly encountered in urate gout, and an upper extremity site of inflammation (wrist, elbow, shoulder) for a first attack should raise suspicion for acute CPP crystal arthritis [13]. These episodes are typically self-limited, and they usually last days to weeks.Trauma, surgery, or severe medical illness often provoke acute attacks. In particular, flares of acute CPP crystal arthritis after parathyroidectomy have been observed [14]; these episodes may be related to abrupt reduction in serum calcium and magnesium levels during postoperative hypoparathyroidism (see "Hungry bone syndrome following parathyroidectomy in end-stage renal disease patients"). Such reduction may cause partial dissolution of crystals with subsequent release from the cartilage matrix into the joint fluid, allowing phagocytosis and the phlogistic response of inflammatory cells.Treatment with pamidronate and other bisphosphonates [15] or granulocyte-macrophage colony-stimulating factor (GM-CSF) [16-18] have also been reported to precipitate acute attacks of pseudogout.A review of 50 cases of acute CPP crystal arthritis, many of which were polyarticular attacks, described low-grade fevers in 50 percent and frequently elevated sedimentation rates [13]. In some cases, systemic features accompanying polyarticular acute CPP crystal arthritis are quite prominent and may suggest pyogenic arthritis, osteomyelitis, and/or systemic sepsis. (See 'Chronic CPP crystal inflammatory arthritis' below.)Rarely, and usually after several acute arthritic flares, palpable and visible masses of CPP crystals, resembling gouty tophi, accumulate in synovium and adjacent joint structures, and may lead to locally destructive and compressive symptoms.Chronic CPP crystal arthritisChronic CPP crystal inflammatory arthritis — The term pseudo-rheumatoid arthritis (pseudo-RA) was applied to a nonerosive, inflammatory arthritis in which CPP crystals were demonstrable in joint fluid [2]. This presentation of CPPD disease resembles RA in several respects, including the presence of significant morning stiffness, fatigue, synovial thickening, localized edema, and restricted joint motion due either to active inflammation or to flexion contracture.Typically, the chronic inflammatory arthritis of CPPD disease involves multiple joints, frequently involving peripheral joints of the upper and lower extremities, including the wrists and metacarpophalangeal (MCP) joints, as well as the knees and elbows, in a symmetric or nearly symmetric pattern. Articular inflammation may last up to several months, and inflammation in affected joints tends to wax and wane independently of one another, in distinction to RA, where synchronous flare and remission are more typical.Chronic CPP crystal inflammatory arthritis occurs in 5 percent or less of patients with symptomatic CPPD disease. A rare subtype of chronic CPP crystal inflammatory arthritis, occurring most often in older adult patients during an acute polyarticular attack, is characterized by prominent systemic features, such as leukocytosis, fever, and mental confusion, closely mimicking systemic sepsis [19]. In such patients, the delirium is reported to resolve with resolution of the acute polyarticular flare. These episodes are typically self-limited, usually lasting from days to weeks.Osteoarthritis with CPPD — This is the most prevalent form of symptomatic CPPD disease; for example, 20 percent of unselected patients examined at total knee joint replacement for OA showed CPP crystals in synovial fluid samples [20]. Approximately 50 percent of patients with symptomatic CPP crystal deposition disease show progressive joint degeneration, usually involving multiple joints. This pattern of disease has historically been referred to as "pseudo-OA" because of its resemblance to OA occurring in the absence of CPPD. In about one-half of such patients, episodes of acute inflammatory arthritis typical of pseudogout punctuate the course. In the remainder, joint degeneration proceeds by a process more typical of classical OA. (See "Clinical manifestations and diagnosis of osteoarthritis".)The most commonly affected joints in this form of CPPD disease are the knees, followed by the wrists, MCP joints, hips, shoulders, elbows, and spine. Although a symmetric pattern of joint involvement is frequent, unilateral or more severe degenerative change on one side is not unusual. Findings on clinical examination of individual joints do not typically differ from those observed in OA, which include asymmetric bony enlargement, tenderness, effusions, crepitus, and restricted joint motion. Patients with OA with CPPD may also exhibit contractures of involved joints and valgus deformities of the knees.The arthritic process of CPPD may occur in joints typically involved in OA, such as the interphalangeal joints of the hands, the first carpometacarpal joints, the knees, or the first metatarsophalangeal joints. An etiologic or accelerating role for CPP crystal deposition in joint degeneration seems likely when radiographic calcification is present early in the course of degeneration, particularly if the degenerative changes also involve joints atypical for OA (wrists, MCP joints, elbows, and shoulders) in the absence of a preceding history of joint trauma or of vocational or avocational stress.As cartilage is lost during the course of progressive joint degeneration, previously apparent cartilage calcification may become increasingly difficult or impossible to detect radiographically, thereby obscuring the diagnosis. (See 'Imaging findings' below.)Severe joint degeneration — A number of reports have documented CPP crystal deposition in association with severe joint degeneration which closely resembles neuropathic arthropathy [21-23]. Neuropathic arthropathy is characterized by severe joint degeneration and disruption occurring in the course of neurologic disorders leading to joint denervation; the affected joint is often called a Charcot joint. Underlying disorders associated with Charcot joints include diabetes mellitus (most common), tabes dorsalis, and syringomyelia. (See "Diabetic neuropathic arthropathy".)In contrast to neuropathic arthropathy, neurologic function is typically normal in severe joint degeneration associated with CPP crystal deposition, prompting use of the term "pseudo-neuropathic joint disease" for this relationship, although an underlying neurologic impairment (typically tabes dorsalis) has, in some instances, been demonstrated [22,23]. The term "pseudo-neuropathic" is intended to convey the view that, when CPP crystal deposition is associated with certain neurologic deficits, the presence of crystals amplifies the destructive consequences of joint denervation.Spinal involvement — CPP crystal deposition in and about the spine has been associated with a number of clinical manifestations, including spine stiffness, sometimes associated with bony ankylosis, which can resemble the spinal changes of ankylosing spondylitis or diffuse idiopathic skeletal hyperostosis (DISH). Such symptoms have been most commonly encountered in familial CPPD disease [24]. In addition, crystal deposition in the ligamentum flavum at the cervical spine level or in the posterior longitudinal ligament at lower levels of the spine may lead to spinal cord compression syndromes or to symptoms either of acute nerve compression or of chronic spinal stenosis [25-27].The crowned dens syndrome (CDS) is a rare, but important to recognize, syndrome, which is characterized by severe acute or recurrent axial neck pain, neck and shoulder girdle stiffness, and associated fever; elevated inflammatory markers (C-reactive protein, erythrocyte sedimentation rate, and CPP or basic calcium phosphate crystal deposition demonstrable on computed tomography [CT] in and around the atlanto-axial articulation) (image 2) [28,29]. The importance of identification of the crystal deposition basis of CDS lies both in the resemblance of its symptoms and signs to those of polymyalgia rheumatica, giant cell arteritis, or, less frequently, meningitis, cervical discitis, or inflammatory spondyloarthritis; and in the usually favorable response of CDS clinical features to treatment with nonsteroidal antiinflammatory drugs (NSAIDs) or colchicine.Other manifestations — CPP crystal deposition occurring in the wake of trauma or prior surgery may result in localized inflammation or degeneration, most commonly in the knee or in the lower or lumbar spine. In addition, CPP crystal deposition in bursae, ligaments, and tendons may be sufficient to cause local nerve compression, as in the carpal tunnel [30].SYNOVIAL FLUID FINDINGS — The most salient finding on synovial fluid analysis in calcium pyrophosphate (CPP) crystal deposition (CPPD) disease is the presence of positively birefringent CPP crystals by compensated polarized light microscopy. In inflamed joints during an attack of acute CPP crystal arthritis, phagocytosed crystals within polymorphonuclear leukocytes are virtually always present (picture 1).Total synovial fluid leukocyte concentration in an acute attack is typically 15,000 to 30,000 per mm3, 90 percent of which are neutrophils. In chronically symptomatic joints, cell counts are typically lower.The abundance of CPP crystals in the synovial fluid is generally related to the degree of clinically apparent inflammation [2]; however, large discrepancies between these variables can occur.CPP crystals differ from the needle-shaped, strongly and negatively birefringent monosodium urate crystals in acute gouty arthritis (picture 2); CPP crystals are more difficult to detect than monosodium urate crystals because they are:●Smaller (0.5 to 10 microns)●Weakly positively birefringent or not birefringent at all●More polymorphic with rod-shaped and cuboid crystals in addition to the usual rhomboidal formIn some instances, CPP crystals are too small to be readily visualized; for example, CPP crystals are sometimes smaller than can be resolved even by 1000-fold magnification with the aid of phase contrast microscopy [31].Patients with CPP crystals in a synovial fluid sample may also have the simultaneous presence of monosodium urate crystals. The coexistence of urate and CPP crystals in a single inflammatory effusion is neither uncommon nor unexpected, given the observed frequencies of hyperuricemia (20 percent) and gout (about 5 percent) among patients with CPPD disease [2].IMAGING FINDINGS — Imaging evidence for calcium pyrophosphate (CPP) crystal deposition (CPPD) has traditionally relied upon conventional radiography, which reveals findings of cartilage calcification (see 'Cartilage calcification (chondrocalcinosis)' below). Degenerative changes in the joint are also frequently present, and certain radiographic findings in particular joints are characteristic of CPPD disease in those locations (see 'Degenerative changes' below and 'Other radiographic features in specific joints' below). Ultrasonographic findings that correlate with radiographic features of CPPD disease have also been described (see 'Ultrasonographic findings' below). Magnetic resonance imaging (MRI) is a less sensitive imaging modality for documenting CPPD than conventional radiography, ultrasonography, or computed tomography (CT).Plain film radiographyCartilage calcification (chondrocalcinosis) — Radiographic evidence of CPPD is the defining feature of this finding. CPPD typically appears as punctate and linear radiodensities in articular cartilage (fibrocartilage and/or hyaline cartilage) (image 1) and, with lesser frequency, in ligaments, tendons, synovia, bursae, and joint capsules. Although deposits of basic calcium phosphate crystals may occasionally cause confusion, such deposits are usually faint and are irregularly contoured.●Cartilage – Among affected fibrocartilages in CPPD disease are the menisci of the knee (usually bilaterally), the symphysis pubis, the triangular fibrocartilage of the wrist joints, and the glenoid and acetabular labra. CPPD in hyaline cartilage frequently appear as a radiopaque line paralleling the surface of the underlying bone.●Joints – Larger joints, such as the knee, wrist, elbow, shoulder, and hip, are most frequently involved in CPPD disease, but almost any diarthrodial joint may be affected radiographically. Articular capsule or synovial calcification is often fainter and more diffuse than cartilage calcification.Linear calcifications involving the Achilles tendon or plantar fascia are often seen in CPPD disease [32].Degenerative changes — CPPD is often associated with degenerative changes in joints, even in the absence of radiographic cartilage calcification. Characteristic degenerative changes in CPPD disease include typical features of osteoarthritis, including subchondral cysts, osteophyte formation, and bone and cartilage fragmentation.CPPD disease may also underlie the occurrence of radiographic features of osteoarthritis (OA) in joints not commonly affected by primary OA (see 'Diagnostic criteria' below and 'Differential diagnosis' below). Cartilage calcification that is apparent at the onset of degenerative changes or which occurs earlier in life than usual and without a pertinent vocational or avocational history is also characteristic of CPPD disease, whether in joints typical or atypical for OA.Other radiographic features in specific joints — A variety of radiographic signs restricted to joints or regions are more or less characteristic of CPPD disease, sometimes in addition to cartilage calcification (see 'Cartilage calcification (chondrocalcinosis)' above). The affected joints and findings include:●Metacarpophalangeal (MCP) joints – Squared-off bone ends and hook-like osteophytes in the MCP joints, particularly if these changes are located in the second and third MCP joints. Such changes are especially common in hemochromatosis [33] and hemochromatosis-associated CPPD disease [34]. (See "Clinical manifestations and diagnosis of hereditary hemochromatosis".)●Wrist – Isolated or unusually extensive radiocarpal joint narrowing and/or navicular-lunate dissociation [35]●Patellofemoral joints – Severe patellofemoral joint space degeneration, especially with a wrapped-around deformity of the patella on the femur. This finding is seen in hyperparathyroidism with or without CPPD. Notching or erosion of the distal femoral cortex superior to the patella may also occur [36].●Spine and pelvis – Axial skeleton changes, such as subchondral cysts in the small joints of the spine and in the sacroiliac joints, calcification of multiple intervertebral discs, and sacroiliac joint vacuum phenomena [37].Crowned dens syndrome (CDS) (see 'Spinal involvement' above) can be identified by use of CT (image 2), which is the preferred modality to demonstrate its presence. Findings may include CPP (or basic calcium phosphate) crystal masses at the atlanto-axial articulation and/or in the transverse ligament of the atlas and/or in the ligamentum flavum [38].Ultrasonographic findings — The following findings on ultrasonography of articular and fibrocartilage may be indicative of the presence of deposits of CPP crystals [39,40]:●A thin hyperechoic band paralleling the bone cortex and separated from it by a hypoechoic region representing cartilage. The resulting ultrasonographic appearance resembles the double contour sign (DCS) initially described in gout [41], but it often exhibits a thin, stippled appearance rather than the smooth pattern characteristic of gout.●Small hyperechoic rounded amorphous shaped regions, often with acoustic shadowing, which are most often found in images of fibrocartilage of the wrist (image 3) and menisci of the knee, and in tendons.●Nodular hyperechoic deposits in bursae and articular recesses.●Hyperechoic lines of calcification running parallel to tendon fibers.In contrast to urate crystal deposits in gout, CPP crystals often deposit within the substance of hyaline cartilage, providing a potentially attractive means to distinguish between these crystal deposition arthropathies.Ultrasonography is a promising modality for clinical use in the diagnosis of CPPD disease and tracking the efficacy of CPPD disease therapies; however, further studies are warranted for: validation of ultrasound criteria unique to CPP crystal deposition, resolution of differences reported with regard to the sensitivity and specificity of the procedure for the diagnosis of CPPD disease [39,42-45], and comparison of imaging findings with corresponding histopathology as the gold standard.Based upon the limited data available, ultrasonography appears to have sensitivity for aspiration-confirmed CPPD disease similar to that of conventional radiography when employed by experts in joint imaging [39]. The utility of ultrasonography in diagnosing cartilage calcification outside of research centers, however, remains uncertain. Thus, we do not believe that the results of ultrasonographic study yet qualify as a diagnostic imaging criterion for CPPD disease. |
|
|
|
60 year old female presented to the OPd with lethargy, LOA,abdominal pain and hopelessness for 2 months. Likely diagnoses include CA stomach Social phobia Depression Schizophrenia Hypercalcaemia |
Q |
|
|
|
Why do we need Nicorandil Is it for secondary prevention |
AbstractNicorandil is a drug with both nitrate-like and ATP-sensitive potassium-channel (K+ ATP) activating properties. By virtue of this dual mechanism of action, the drug acts as a balanced coronary and peripheral vasodilator and reduces both preload and afterload. The K+ ATP channel has been shown to be involved in the phenomenon of myocardial preconditioning, and studies in animal models of ischaemia-reperfusion-induced myocardial stunning or infarction indicate that nicorandil has cardio-protective effects. Studies in patients undergoing percutaneous transluminal coronary angioplasty (PTCA) have shown that the administration of nicorandil reduces ST-segment elevation during ischaemia. Nicorandil significantly improved the results of exercise tolerance tests versus baseline in patients with stable effort angina pectoris in early noncomparative trials. The drug also improved the results of exercise tolerance tests relative to placebo in early randomised, double-blind, placebo-controlled trials. In randomised, double-blind comparative studies in patients with angina pectoris, nicorandil has demonstrated equivalent efficacy, as measured by exercise tolerance testing, to isosorbide di- and mononitrate, metoprolol, propranolol, atenolol, diltiazem, amlodipine and nifedipine. The effects of nicorandil on various aspects of myocardial recovery from ischaemic damage caused by acute myocardial infarction have been investigated in the short term. Regional left ventricular (LV) wall motion, a marker of myocardial function, was significantly improved in nicorandil recipients relative to control. The main adverse event associated with nicorandil as treatment for angina pectoris is headache. This can be minimised by commencing nicorandil at a low dose in patients prone to headache. There have been infrequent case reports of mouth ulcers in patients receiving nicorandil; causality has not been conclusively established, but product prescribing information indicates that an alternative treatment should be considered if persistent aphthous or severe mouth ulceration occurs. Thus, nicorandil remains a useful background therapy for patients with angina pectoris. The drug has also demonstrated potential cardioprotective effects when used as part of an intervention strategy directly after acute myocardial infarction in high-risk patients. Further large scale longer term studies of nicorandil in this latter indication are awaited with interest. |
|
|
|
36. Coronary angioplasty and stenting Performed more often than surgery in many centres Diabetics are a subgroup with best outcome from angioplasty than surgery Similar outcome to surgery in 3 vessel disease but at the expense of repeat procedures Primary angioplasty is a better alternative than thrombolytics in experienced centres Anti neoplastics are used in cardiac stents that reduce restenosis |
Q CABG |
|
|
|
What |
Q |
|
|
|
Raloxifene on oestrogen receptors Activation or modulation |
Q |
|
|
|
Calcitonin mode of administration |
IV / IM / SC |
|
|
|
Which medications have not shown conclusively to improve prognosis post MIClopidogrelCCBLMWHNicorandilPolyunsaturated fat fish oil
|
This site is intended for healthcare professionalsMedscape LogoDrugs & Diseases > CardiologyMyocardial Infarction MedicationUpdated: Jul 19, 2018 Author: A Maziar Zafari, MD, PhD; Chief Editor: Eric H Yang, MD more...Share FeedbackSECTIONSMedication SummaryThe goals of pharmacotherapy for myocardial infarction are to reduce morbidity and to prevent complications. The main goals of emergency department medical therapy are rapid intravenous thrombolysis and/or rapid referral for percutaneous coronary intervention (PCI), optimization of oxygenation, reduction of cardiac workload, and pain control. Clinicians may consider titrated intravenous opioids for analgesia or a mild tranquilizer (eg, a benzodiazepine) for very anxious patients. [70]Antiplatelet AgentsClass SummaryAntiplatelet agents have a strong mortality benefit. There is an increased risk of bleeding in cases of emergency coronary artery bypass graft (CABG).Aspirin (Ascriptin, Bayer Aspirin, Aspirtab, Ecotrin, Durlaza)View full drug informationEarly administration of aspirin in patients with acute myocardial infarction has been shown to reduce cardiac mortality rate by 23% in the first month.Clopidogrel (Plavix)View full drug informationClopidogrel selectively inhibits adenosine diphosphate (ADP) binding to platelet receptors and subsequent ADP-mediated activation of glycoprotein GPIIb/IIIa complex, thereby inhibiting platelet aggregation.Clopidogrel may have a positive influence on several hemorrhagic parameters and may exert protection against atherosclerosis, not only through inhibition of platelet function but also through changes in the hemorrhagic profile.This agent has been shown to decrease cardiovascular death, myocardial infarction, and stroke in patients with acute coronary syndrome (ie, unstable angina, non-ST elevation MI [NSTEMI], or ST-elevation MI [STEMI]).Ticagrelor (Brilinta)View full drug informationTicagrelor and its major metabolite reversibly interact with the platelet P2Y12 ADP-receptor to prevent signal transduction and platelet activation. This agent is indicated to reduce the rate of thrombotic cardiovascular events in patients with acute coronary syndrome (ACS)—that is, unstable angina, non-ST elevation MI (NSTEMI), or ST-elevation MI (STEMI). Ticagrelor also reduces the rate of stent thrombosis in patients who have undergone stent placement for treatment of ACS, and it is indicated in patients with a history of MI more than 1 year previously. Patients can be transitioned from clopidogrel to ticagrelor without interruption of antiplatelet effect.Prasugrel (Effient)View full drug informationPrasugrel is a prodrug, a thienopyridine that inhibits platelet activation and aggregation through irreversible binding of active metabolite to adenosine phosphate (ADP) platelet receptors (specifically, P2Y12 receptor)It is indicated for reduction of thrombotic cardiovascular events (including stent thrombosis) in patients with acute coronary syndrome (ACS) managed by means of percutaneous coronary intervention (PCI) who have either (a) unstable angina or non-ST-elevation MI (NSTEMI) or (b) ST-elevation MI (STEMI) when managed with primary or delayed PCI.The use of prasugrel is not recommended for patients with a history of stroke or transient ischemic attack (TIA).Vorapaxar (Zontivity)View full drug informationVorapaxar reversibly inhibits protease-activated receptor 1 (PAR-1) which is expressed on platelets, but its long half-life makes it effectively irreversible. It is indicated to reduce thrombotic cardiovascular events in patients with a history of MI or with peripheral arterial disease. It is not used as monotherapy, but added to aspirin and/or clopidogrel.Antithrombotic AgentsClass SummaryAntithrombotic agents, which include heparin, bivalirudin, and enoxaparin, prevent the formation of thrombi associated with myocardial infarction and inhibit platelet function by blocking cyclooxygenase and subsequent platelet aggregation. Antiplatelet therapy has been shown to reduce mortality rates by reducing the risk of fatal myocardial infarctions, fatal strokes, and vascular death. Unfractionated intravenous heparin and fractionated low-molecular-weight subcutaneous heparins are the 2 choices for initial anticoagulation therapy.Bivalirudin (Angiomax)View full drug informationBivalirudin, a synthetic analogue of recombinant hirudin, inhibits thrombin; it is used for anticoagulation in patients with unstable angina who are undergoing PCI. With provisional use of glycoprotein IIb/IIIa inhibitor (GP IIb/IIIa inhibitor), bivalirudin is indicated for use as an anticoagulant in patients undergoing PCI. Potential advantages over conventional heparin therapy include more predictable and precise levels of anticoagulation, activity against clot-bound thrombin, absence of natural inhibitors (eg, platelet factor 4, heparinase), and continued efficacy following clearance from plasma (because of binding to thrombin).HeparinView full drug informationHeparin augments the activity of antithrombin III and prevents the conversion of fibrinogen to fibrin. Heparin does not actively lyse, but it is able to inhibit further thrombus formation and prevents reaccumulation of a clot after spontaneous fibrinolysis.Enoxaparin (Lovenox)View full drug informationEnoxaparin enhances the inhibition of factor Xa and thrombin by increasing antithrombin III activity. In addition, it preferentially increases the inhibition of factor Xa. Enoxaparin is indicated for the treatment of acute STEMI managed medically or with subsequent PCI. It is also indicated for prophylaxis of ischemic complications caused by unstable angina and non-Q-wave myocardial infarction.Dalteparin (Fragmin)View full drug informationEnhances inhibition of factor Xa and thrombin by increasing antithrombin III activity. In addition, preferentially increases inhibition of factor Xa.Except in overdoses, no utility exists in checking PT or aPTT, because aPTT does not correlate with anticoagulant effect of fractionated LMWH.Average duration of treatment is 7-14 d.Glycoprotein IIb/IIIa InhibitorsClass SummaryGlycoprotein IIb/IIIa inhibitors prevent acute cardiac ischemic complications in unstable angina that is unresponsive to conventional therapy.Abciximab (ReoPro)View full drug informationAbciximab is a chimeric human-murine monoclonal antibody. It binds to the platelet surface glycoprotein IIb/IIIa (GPIIb/IIIa) receptor with high affinity, preventing the binding of fibrinogen and reducing platelet aggregation by 80%. Inhibition of platelet aggregation persists for as long as 48 hours after infusion stops.Tirofiban (Aggrastat)View full drug informationTirofiban is a nonpeptide antagonist of the glycoprotein IIb/IIIa receptor. It is a reversible antagonist of fibrinogen binding, and when administered intravenously, it inhibits platelet aggregation by more than 90%.Eptifibatide (Integrilin)View full drug informationEptifibatide is a cyclic peptide that also reversibly inhibits platelet aggregation by binding to the glycoprotein IIb/IIIa receptor. Blocks platelet aggregation and prevents thrombosis.VasodilatorsClass SummaryVasodilators relieve chest discomfort by improving myocardial oxygen supply, which in turn dilates epicardial and collateral vessels, improving blood supply to the ischemic myocardium.Nitroglycerin IV (Nitro-Dur)View full drug informationNitroglycerin relaxes vascular smooth muscle via stimulation of intracellular cyclic guanosine monophosphate production, causing a decrease in blood pressure. Nitrates are useful for preload reduction and symptomatic relief but have no apparent impact on mortality rate in myocardial infarction.Beta-adrenergic blockersClass SummaryThis category of drugs has the potential to suppress ventricular ectopy due to ischemia or excess catecholamines. In the setting of myocardial ischemia, beta-blockers have antiarrhythmic properties and reduce myocardial oxygen demand secondary to elevations in heart rate and inotropy.Metoprolol (Lopressor)View full drug informationThis category of drugs, which includes metoprolol (Lopressor) and esmolol (Brevibloc), has the potential to suppress ventricular ectopy due to ischemia or excess catecholamines. In the setting of myocardial ischemia, beta-blockers have antiarrhythmic properties and reduce myocardial oxygen demand secondary to elevations in heart rate and inotropy.Esmolol (Brevibloc)View full drug informationEsmolol is a useful drug for patients at risk of experiencing complications from beta-blockers, particularly reactive airway disease, mild-to-moderate left ventricular dysfunction, and peripheral vascular disease. Its short half-life of 8 minutes allows for titration to desired effect, with the ability to stop quickly if necessary.Atenolol (Tenormin)View full drug informationUsed to treat hypertension. Selectively blocks beta1-receptors with little or no effect on beta 2 types. Beta-adrenergic blocking agents affect blood pressure via multiple mechanisms. Actions include negative chronotropic effect that decreases heart rate at rest and after exercise, negative inotropic effect that decreases cardiac output, reduction of sympathetic outflow from the CNS, and suppression of renin release from the kidneys. Used to improve and preserve hemodynamic status by acting on myocardial contractility, reducing congestion, and decreasing myocardial energy expenditure.Beta-adrenergic blockers reduce inotropic state of left ventricle, decrease diastolic dysfunction, and increase LV compliance, thereby reducing pressure gradient across LV outflow tract. Decreases myocardial oxygen consumption, thereby reducing myocardial ischemia potential. Decreases heart rate, thus reducing myocardial oxygen consumption and reducing myocardial ischemia potential.During IV administration, carefully monitor blood pressure, heart rate, and ECG.Angiotensin-Converting Enzyme InhibitorsClass SummaryACE inhibitors may prevent the conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, resulting in lower aldosterone secretion. ACE inhibitors reduce mortality rates after myocardial infarction. Administer ACE inhibitors as soon as possible as long as the patient has no contraindications and remains in stable condition. ACE inhibitors have the greatest benefit in patients with ventricular dysfunction.CaptoprilView full drug informationCaptopril has a short half-life, which makes it an important drug for initiation of ACE inhibitor therapy. It can be started at a low dose and titrated upward as needed and as the patient tolerates.Enalapril (Vasotec, Epaned)View full drug informationEnalapril prevents conversion of angiotensin I to angiotensin II, resulting in increased levels of plasma renin and a reduction in aldosterone secretion. Has a favorable clinical effect when administered over a long period of time.Quinapril (Accupril)View full drug informationQuinapril prevents conversion of angiotensin I to angiotensin II, resulting in increased levels of plasma renin and a reduction in aldosterone secretion.Lisinopril (Zestril, Prinivil)View full drug informationPrevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, resulting in lower aldosterone secretion.Angiotensin-Receptor BlockersClass SummaryAngiotensin-receptor blockers may be used as an alternative to ACE inhibitors in patients who develop adverse effects, such as a persistent cough, although initial trials need to be confirmed. An angiotensin-receptor blocker should be administered to patients with STEMI who are intolerant of ACE inhibitors and who have either clinical or radiologic signs of heart failure or an LVEF of less than 40%.Irbesartan (Avapro)View full drug informationBlocks vasoconstrictor and aldosterone-secreting effects of angiotensin II at tissue receptor site. May induce more complete inhibition of renin-angiotensin system than ACE inhibitors and does not affect response to bradykinin (less likely to be associated with cough and angioedema).Candesartan (Atacand)View full drug informationCandesartan blocks vasoconstriction and aldosterone-secreting effects of angiotensin II. May induce more complete inhibition of renin-angiotensin system than ACE inhibitors, does not affect response to bradykinin, and is less likely to be associated with cough and angioedema. Use in patients unable to tolerate ACE inhibitors.Valsartan (Diovan)View full drug informationProduces direct antagonism of angiotensin II receptors. Displaces angiotensin II from AT1 receptor and may lower blood pressure by antagonizing AT1-induced vasoconstriction, aldosterone release, catecholamine release, arginine vasopressin release, water intake, and hypertrophic responses. Use in patients unable to tolerate ACE inhibitors.Azilsartan (Edarbi)View full drug informationAngiotensin II blocker; displaces angiotensin II from AT1 receptor and may lower blood pressure by antagonizing AT1-induced vasoconstriction, aldosterone release, catecholamine release, arginine vasopressin release, water absorption, and hypertrophic responsesMay induce more complete inhibition of renin-angiotensin system compared with ACE inhibitors; does not affect response to bradykininInhibits the pressor effects of an angiotensin II infusion in a dose-related mannerEprosartan mesylate (Teveten)View full drug informationNonpeptide angiotensin II receptor antagonist that blocks vasoconstrictor and aldosterone-secreting effects of angiotensin II. May induce more complete inhibition of renin-angiotensin system than ACE inhibitors and does not affect response to bradykinin and is less likely to be associated with cough and angioedema.For patients unable to tolerate ACE inhibitors.Angiotensin II receptor blockers reduce blood pressure and proteinuria, protecting renal function, and delaying onset of end-stage renal disease.Losartan (Cozaar)View full drug informationAngiotensin II receptor antagonist that blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II. May induce a more complete inhibition of the renin-angiotensin system than ACE inhibitors, does not affect the response to bradykinin, and is less likely to be associated with cough and angioedema. For patients unable to tolerate ACE inhibitors.ThrombolyticsClass SummaryThe main objective of thrombolysis is to restore circulation through a previously occluded vessel by the rapid and complete removal of a pathologic intraluminal thrombus or embolus that has not been dissolved by the endogenous fibrinolytic system.The first generation of fibrinolytic drugs (eg, streptokinase, urokinase, acetylated plasminogen streptokinase activator complexes [APSACs], reteplase, and novel plasminogen activator [nPA]) indiscriminately induced activation of circulating plasminogen and clot-associated plasminogen. First-generation drugs invariably elicited a systemic lytic state characterized by depletion of circulating fibrinogen, plasminogen, and hemostatic proteins and by marked elevation of concentrations of fibrinogen degradation products in plasma.Second-generation drugs (eg, alteplase [t-PA], single-chain urokinase plasminogen activator), such as tenecteplase, preferentially activate plasminogen in the fibrin domain, rather than in the circulation, as with free plasminogen. Therefore, they have clot selectivity. Tenecteplase should be initiated as soon as possible in patients with STEMI; tenecteplase is administered as a single bolus, exhibiting a biphasic disposition from the plasma.In optimal regimens, these agents induce clot lysis without inducing a systemic lytic state, they are less prone than nonselective agents to predispose the patient to hemorrhage necessitating transfusion, and they are effective in inducing recanalization in 80-90% of infarct-related arteries within 90 minutes. Therefore, t-PA recanalizes 75-80% of infarct-related arteries.Alteplase, t-PA (Activase)View full drug informationAlteplase (t-PA) is a fibrin-specific agent with a brief half-life of 5 minutes. Adjunctive therapy with IV heparin is necessary to maintain the patency of arteries recanalized by t-PA, especially during the first 24-48 hours.Tenecteplase (TNKase)View full drug informationTenecteplase is a modified version of alteplase (t-PA) made by substituting 3 amino acids of alteplase. It can be given as a single bolus over a 5-second infusion, instead of 90 minutes with alteplase. Tenecteplase appears to cause less nonintracranial bleeding, but the risk of intracranial bleeding and stroke is similar to that of alteplase. Base the dose using patient weight. Initiate treatment as soon as possible after the onset of acute STEMI symptoms. Because tenecteplase contains no antibacterial preservatives, reconstitute immediately before use.AnalgesicsClass SummaryPain control is essential to quality patient care. Analgesics ensure patient comfort, promote pulmonary toilet, and have sedating properties, which are beneficial for patients who experience pain.Morphine sulfate (Duramorph, MS Contin, Kadian, Avinza)View full drug informationMorphine sulfate is the drug of choice for narcotic analgesia due to its reliable and predictable effects, safety profile, and ease of reversibility with naloxone. Morphine sulfate is administered intravenously, may be dosed in a number of ways, and commonly is titrated until the desired effect is achieved.PCSK9 InhibitorsClass SummaryOver the last decade, inhibition of proprotein convertase subtilisin/kexin type 9 (PCSK9) has emerged as a promising target to reduce residual cardiovascular disease risk. PCSK9 is a protein that binds to low-density lipoprotein (LDL) receptors (LDLR) to promote their degradation. Monoclonal antibodies inhibit PCSK9 and thus prevent LDLR degradation. This action will increase the number of LDLRs and subsequently increase the clearance of LDL, ultimately lowering LDL-C levels. In December 2017, the FDA approved the first PCSK9 inhibitor, evolocumab (Repatha), for the prevention of strokes, heart attacks, and coronary revascularizations. [64] The approval was based on data from the evolocumab cardiovascular outcomes study (FOURIER). In the FOURIER clinical trial, evolocumab demonstrated significant benefits for 27,564 patients with established cardiovascular disease. The study revealed that when used in addition to optimized statin therapy, evolocumab reduced the risk of heart attack by 27%, the risk of stroke by 21%, and the risk of coronary revascularization by 22%. In addition, evolocumab showed a statistically significant 15% reduction in the risk of the primary composite endpoint, which included hospitalization for unstable angina, coronary revascularization, heart attack, stroke, or cardiovascular death. [65]Evolocumab (Repatha)View full drug informationHuman monoclonal IgG2 directed against PCSK9. Evolocumab binds to PCSK9 and inhibits circulating PCSK9 from binding to the LDLR, preventing PCSK9-mediated LDLR degradation and permitting LDLR to recycle back to the liver cell surface. By inhibiting the binding of PCSK9 to LDLR, evolocumab increases the number of LDLRs available to clear LDL from the blood, thereby lowering LDL-C levels. eMedicine LogoQuestionsSECTIONSMyocardial InfarctionOverviewPresentationDDxWorkupTreatmentGuidelinesMedicationMedication SummaryAntiplatelet AgentsAntithrombotic AgentsGlycoprotein IIb/IIIa InhibitorsVasodilatorsBeta-adrenergic blockersAngiotensin-Converting Enzyme InhibitorsAngiotensin-Receptor BlockersThrombolyticsAnalgesicsPCSK9 InhibitorsQuestions & AnswersMedia GalleryTablesReferencesWhat to Read Next on MedscapeRelated Conditions and DiseasesFast Five Quiz: Are You Prepared to Confront a Myocardial Infarction?Pathology of Acute Myocardial InfarctionThrombolysis in Myocardial Infarction (TIMI) ScoreFast Five Quiz: Can You Recognize Subtle and Surprising Signs of Heart Conditions?Acute Myocardial Infarct ImagingRight Ventricular InfarctionNEWS & PERSPECTIVE SCAD: Spontaneous Healing Common With Conservative TreatmentBroken PROMISE: CTA for Suspected CAD in Patients With DiabetesWhy Women's Cardiovascular Health Needs More AttentionTOOLSDrug Interaction CheckerPill IdentifierCalculatorsFormularySLIDESHOW Top News From ESC 2017: SlideshowMost Popular ArticlesAccording to Cardiologists Alcohol and the Electric Atrium: How Drinking Promotes AF? Marijuana Edible Linked to Myocardial Infarction High Fiber, Whole Grains Linked to Lower CVD, Diabetes, Cancer Risk FDA Warns of Aortic Aneurysm Risk With Fluoroquinolones Nutrition Research: Restoring Respect to a Field Under SiegeView More Medscape LogoFIND US ON ABOUTAbout MedscapePrivacy PolicyCookiesTerms of UseAdvertising PolicyHelp CenterMEMBERSHIPEmail NewslettersManage My AccountAPPSMedscapeMedPulse NewsCME & EducationWEBMD NETWORKWebMDMedicineNeteMedicineHealthRxListWebMD CorporateEDITIONSEnglishDeutschEspañolFrançaisPortuguêsAll material on this website is protected by copyright, Copyright © 1994-2019 by WebMD LLC. This website also contains material copyrighted by 3rd parties. |
|
|
|
Essential tremor Autosomal recessive or what |
Q |
|
|
|
HCT CAUSE HYPERCHOLESTEROLEMIA AND HYPERLIPIDEMIA BUT DOES IT CAUSE HYPERTRIGLYCERIDAEMIA |
Q |
|
|
|
Q. DonepezilIs a reversible cholinesterase inhibitorSlow the progression of cognitive decline in AlzheimersMay cause vivid dreamsDrug holidays are advised when used long termCan cause hepatotoxicity
|
Mechanism of ActionAcetylcholinesterase inhibitor that causes an increase in concentrations of acetylcholine, which in turn enhances cholinergic neurotransmissionAbsorptionBioavailability: 100%Peak plasma time: 3-4 hrDistributionProtein bound: 96%Vd: 12-16 L/kgMetabolismHepatic P-450 enzymes CYP2D6, CYP3A4Metabolites: 4 major metabolites, 2 activeEliminationHalf-life: 70 hrTotal body clearance: 0.13 L/hr/kgExcretion: Urine (57%), feces (17%) Donepezil (Aricept), is a centrally acting reversible acetyl cholinesterase inhibitor. Its main therapeutic use is in the treatment of Alzheimer's disease where it is used to increase cortical acetylcholine. Donepezil is postulated to exert its therapeutic effect by enhancing cholinergic function. This is accomplished by increasing the concentration of acetylcholine through reversible inhibition of its hydrolysis by acetylcholinesterase. If this proposed mechanism of action is correct, donepezil's effect may lessen as the disease process advances and fewer cholinergic neurons remain functionally intact. Donepezil has been tested in other cognitive disorders including Lewy body dementia and Vascular dementia, but it is not currently approved for these indications. Donepezil has also been studied in patients with Mild Cognitive Impairment, schizophrenia, attention deficit disorder, post-coronary bypass cognitive impairment, cognitive impairment associated with multiple sclerosis, and Down syndrome. |
|
|
|
Does parathyroid hormone cause Hypercalciurea |
Q |
|
|
|
Drugs to discharge patients with following MI |
Q |
|
|
|
Q indications for the use of ACE inhibitors Essential HT Micoalbuminuria in DM in the absence of HT💡 Primary pulmonary Ht Portal Ht CCF Is it for microalbuminuria |
ACEI USES |
|
|
|
Delirium tremens |
Delirium tremens usually begins 48 to 72 h after alcohol withdrawal; anxiety attacks, increasing confusion, poor sleep (with frightening dreams or nocturnal illusions), profuse sweating, and severe depression also occur. Fleeting hallucinations that arouse restlessness, fear, and even terror are common. Typical of the initial delirious, confused, and disoriented state is a return to a habitual activity; eg, patients frequently imagine that they are back at work and attempt to do some related activity. Autonomic lability, evidenced by diaphoresis and increased pulse rate and temperature, accompanies the delirium and progresses with it. Mild delirium is usually accompanied by marked diaphoresis, a pulse rate of 100 to 120 beats/min, and a temperature of 37.2 to 37.8° C. Marked delirium, with gross disorientation and cognitive disruption, is accompanied by significant restlessness, a pulse of > 120 beats/min, and a temperature of > 37.8° C; risk of death is high. During delirium tremens, patients are suggestible to many sensory stimuli, particularly to objects seen in dim light. Vestibular disturbances may cause them to believe that the floor is moving, the walls are falling, or the room is rotating. As the delirium progresses, resting tremor of the hand develops, sometimes extending to the head and . aAtaxia is marked; care must be taken to prevent self-injury. Symptoms vary among patients but are usually the same for a particular patient with each recurrence. |
|
|
|
Can bisphosphonates inhibit osteoclasts immediately Which one |
Q |
|
|
|
Are bisphosphonates irreversibly bound to hydroxyappatite |
Q |
|
|
|
Q 47.Drugs which are used in the Rx of glucocorticoid induced osteoporosis includeAlendronateResidronateRaloxifeneStrontium ranelateHRt
|
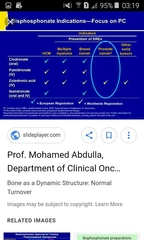
As a result of negative calcium balance and inhibited osteoblast activity, bone tissue deminaralization foci occur. |
|
|
|
What |
Q |
|
|
|
What are the factors affecting drug metabolism |
Q |
|
|
|
Can there be fasciculations without muscle atrophy in AML |
Fasciculation without weakness, muscle atrophy or increased tendon reflexes suggests a benign fasciculation syndrome, even when of sudden onset. Regardless of origin, fasciculations often present as the initial abnormality in ALS, an early harbinger of dysfunction and aberrant firing of motor neurons. |
|
|
|
What is the median age of onset of AML |
Q |
|
|
|
What are the systemic manifestations of RA |
? Eccrine hydradenitis ? Erythema induratum |
|
|
|
Systemic manifestations of copd |
Heart failure ? Cognitive impairment |
|
|
|
. 65 yr old faremer with a hx of smoking 25 pack years presents with progressive SOB.O/E of the chest fine crepts in the lowr zone of the lungs were noted.WOF are T/F regarding this pt?
An HRCT of this pt may reveal honeycombing
The reasoen for fine crepts is traction bronchiectasis
Presence of digital clubbing will be more suggestive of Idiopathic pulmonary fibrosis
Him stating that he has joint pains has no relationship to this current presentation
The fact that he is a farmer should be further looked into facilitate a diagnosis
|
Q Conditions causing fine crepitations
What causes honey combing
What is farmers lung |
Crackles are defined as a short, explosive, nonmusical sound They can be divided into two types: fine and coarse.
Compared with coarse crackles, fine crackles have a higher frequency and a shorter duration.
Fine crackles are caused by the sudden opening of a closed airway;
coarse crackles are thought to related to secretions. Crackles may occur on either inspiration or expiration but are more common during inspiration. Inspiratory crackles may be classified as early inspiratory, midinspiratory, or late inspiratory.
Crackles are more frequently heard in the basilar regions of the lungs because the distribution of airway closure is gravity-dependent.
The number of crackles has been shown to correlate with disease severity.
Crackles may be heard in cardiac disease, fibrotic lung disease, obstructive lung disease, and pulmonary infections.
They may also be heard in healthy older individuals
General characteristics of these crackles have been described for many different disorders (though there may be variations among individual patients).
In idiopathic pulmonary fibrosis, crackles have been described as fine, short in duration, higher-pitched, and occurring in late inspiration. A basilar predominance exists in early disease.
Asbestosis is associated with fine crackles.
The presence of crackles has been shown to be associated with honeycombing on imaging and with the duration of dust exposure.
In bronchiectasis, crackles have been described as high-frequency and coarse. They occur in early inspiration or midinspiration and are thought to be secondary to bronchial wall collapse during expiration and sudden opening in inspiration.
In COPD, crackles are most commonly due to airway secretions and typically disappear after coughing; they may also be due to the opening and closing of narrowed bronchi with weakened airway walls. Crackles in COPD are characterized as coarse, early, and low pitched and tend to be infrequent.
The crackles associated with pulmonary edema are attributed to the opening of airways narrowed by peribronchial edema. They are described as coarse, late-occurring, and high-pitched. They may be inspiratory or expiratory.
In pneumonia, two types of crackles have been described. Early pneumonia is associated with coarse, midinspiratory crackles; crackles during the recovery phase are described as shorter and occurring at the end of expiration.
Crackles are relatively rare with sarcoidosis (because of the upper-lobe predominance of the disease); when they do occur, they are described as fine and either late inspiratory or midinspiratory. |
|
|
What causes clubbing It's present in CHRONIC PULMONARY CONDITIONS |
LUNG CARCINOMA - most commonly Bronchiectasis Cystic fibrosis Idiopathic pulmonary fibrosis Cyanotic heart disease cirrhosis Infective endocarditis inflammatory bowel disease Stroke |
|
|
|
Systolic vs Diastolic heart failure |
Heart failure with reduced ejection fraction (HFrEF)In HFrEF (also called systolic HF), global LV systolic dysfunction predominates. The LV contracts poorly and empties inadequately, leading to increased diastolic volume and pressure and decreased ejection fraction. Many defects in energy utilization, energy supply, electrophysiologic functions, and contractile element interaction occur, with abnormalities in intracellular calcium modulation and cAMP production.Predominant systolic dysfunction is common in heat failure due to myocardial infarction, myocarditis, and dilated cardiomyopathy. Systolic dysfunction may affect primarily the LV or the right ventricle (RV); LV failure often leads to RV failure.Heart failure with preserved ejection fraction (HFpEF)In HFpEF (also known as diastolic heart failure), LV filling is impaired, resulting in increased LV end-diastolic pressure at rest or during exertion. Global contractility and hence ejection fraction remain normal. In most patients with HFpEF, LV end-diastolic volume is normal. However, in some patients, marked restriction to LV filling can cause inappropriately low LV end-diastolic volume and thus cause low CO and systemic symptoms. Elevated left atrial pressures can cause pulmonary hypertension and pulmonary congestion.Diastolic dysfunction usually results from impaired ventricular relaxation (an active process), increased ventricular stiffness, valvular disease, or constrictive pericarditis. Acute myocardial ischemia is also a cause of diastolic dysfunction. Resistance to filling increases with age, reflecting both cardiomyocyte dysfunction and cardiomyocyte loss, and increased interstitial collagen deposition; thus, diastolic dysfunction is particularly common among the elderly. Diastolic dysfunction predominates in hypertrophic cardiomyopathy, disorders with ventricular hypertrophy (eg, hypertension, significant aortic stenosis), and amyloid infiltration of the myocardium. LV filling and function may also be impaired if marked increases in RV pressure shift the interventricular septum to the left.Diastolic dysfunction has increasingly been recognized as a cause of HF. Estimates vary, but about 50% of patients with heart failure have HFpEF; the prevalence increases with age and in patients with diabetes. It is now known that HFpEF is a complex, heterogenous, multiorgan, systemic syndrome, often with multiple concomitant pathophysiologies. Current data suggest that multiple comorbidities (eg, obesity, hypertension, diabetes, chronic kidney disease) lead to systemic inflammation, widespread endothelial dysfunction, cardiac microvascular dysfunction, and, ultimately, molecular changes in the heart that cause increased myocardial fibrosis and ventricular stiffening. Thus, although HFrEF is typically associated with primary myocardial injury, HFpEF may be associated with secondary myocardial injury due to abnormalities in the periphery.LV failureIn heart failure due to left ventricular dysfunction, CO decreases and pulmonary venous pressure increases. When pulmonary capillary pressure exceeds the oncotic pressure of plasma proteins (about 24 mm Hg), fluid extravasates from the capillaries into the interstitial space and alveoli, reducing pulmonary compliance and increasing the work of breathing. Lymphatic drainage increases but cannot compensate for the increase in pulmonary fluid. Marked fluid accumulation in alveoli (pulmonary edema) significantly alters ventilation-perfusion (V/Q) relationships: Deoxygenated pulmonary arterial blood passes through poorly ventilated alveoli, decreasing systemic arterial oxygenation (Pao2) and causing dyspnea. However, dyspnea may occur before V/Q abnormalities, probably because of elevated pulmonary venous pressure and increased work of breathing; the precise mechanism is unclear.In severe or chronic LV failure, pleural effusions characteristically develop, further aggravating dyspnea. Minute ventilation increases; thus, Paco2 decreases and blood pH increases (respiratory alkalosis). Marked interstitial edema of the small airways may interfere with ventilation, elevating Paco2—a sign of impending respiratory failure.RV failureIn heart failure due to right ventricular dysfunction, systemic venous pressure increases, causing fluid extravasation and consequent edema, primarily in dependent tissues (feet and ankles of ambulatory patients) and abdominal viscera. The liver is most severely affected, but the stomach and intestine also become congested; fluid accumulation in the peritoneal cavity (ascites) can occur. RV failure commonly causes moderate hepatic dysfunction, with usually modest increases in conjugated and unconjugated bilirubin, PT, and hepatic enzymes (particularly alkaline phosphatase and gamma-glutamyl transpeptidase [GGT]). The impaired liver breaks down less aldosterone, further contributing to fluid accumulation. Chronic venous congestion in the viscera can cause anorexia, malabsorption of nutrients and drugs, protein-losing enteropathy (characterized by diarrhea and marked hypoalbuminemia), chronic GI blood loss, and rarely ischemic bowel infarction.Cardiac responseIn HFrEF, left ventricular systolic function is grossly impaired; therefore, a higher preload is required to maintain CO. As a result, the ventricles are remodeled over time: The LV becomes less ovoid and more spherical, dilates, and hypertrophies; the RV dilates and may hypertrophy. Initially compensatory, these changes eventually increase diastolic stiffness and wall tension (ie, diastolic dysfunction develops), compromising cardiac performance, especially during physical stress. Increased wall stress raises oxygen demand and accelerates apoptosis (programmed cell death) of myocardial cells. Dilation of the ventricles can also cause mitral or tricuspid valve regurgitation (due to annular dilation) with further increases in end-diastolic volumes.Hemodynamic responsesWith reduced CO, oxygen delivery to the tissues is maintained by increasing oxygen extraction from the blood and sometimes shifting the oxyhemoglobin dissociation curve (see Figure: Oxyhemoglobin dissociation curve.) to the right to favor oxygen release.Reduced CO with lower systemic blood pressure activates arterial baroreflexes, increasing sympathetic tone and decreasing parasympathetic tone. As a result, heart rate and myocardial contractility increase, arterioles in selected vascular beds constrict, venoconstriction occurs, and sodium and water are retained. These changes compensate for reduced ventricular performance and help maintain hemodynamic homeostasis in the early stages of heart failure. However, these compensatory changes increase cardiac work, preload, and afterload; reduce coronary and renal perfusion; cause fluid accumulation resulting in congestion; increase potassium excretion; and may cause cardiomyocyte necrosis and arrhythmias.Renal responsesAs cardiac function deteriorates, renal blood flow decreases (due to low cardiac output). In addition, renal venous pressures increase, leading to renal venous congestion. These changes both result in a decrease in GFR, and blood flow within the kidneys is redistributed. The filtration fraction and filtered sodium decrease, but tubular resorption increases, leading to sodium and water retention. Blood flow is further redistributed away from the kidneys during exercise, but renal blood flow improves during rest.Decreased perfusion of the kidneys (and possibly decreased arterial systolic stretch secondary to declining ventricular function) activates the renin-angiotensin-aldosterone system (RAAS), increasing sodium and water retention and renal and peripheral vascular tone. These effects are amplified by the intense sympathetic activation accompanying heart failure.The renin-angiotensin-aldosterone- vasopressin (antidiuretic hormone [ADH]) system causes a cascade of potentially deleterious long-term effects. Angiotensin II worsens HF by causing vasoconstriction, including efferent renal vasoconstriction, and by increasing aldosterone production, which enhances sodium reabsorption in the distal nephron and also causes myocardial and vascular collagen deposition and fibrosis. Angiotensin II increases norepinephrine release, stimulates release of vasopressin, and triggers apoptosis. Angiotensin II may be involved in vascular and myocardial hypertrophy, thus contributing to the remodeling of the heart and peripheral vasculature, potentially worsening HF. Aldosterone can be synthesized in the heart and vasculature independently of angiotensin II (perhaps mediated by corticotropin, nitric oxide, free radicals, and other stimuli) and may have deleterious effects in these organs.Heart failure that causes progressive renal dysfunction (including that renal dysfunction caused by drugs used to treat HF) contributes to worsening HF and has been termed the cardiorenal syndrome.Neurohumoral responsesIn conditions of stress, neurohumoral responses help increase heart function and maintain BP and organ perfusion, but chronic activation of these responses is detrimental to the normal balance between myocardial-stimulating and vasoconstricting hormones and between myocardial-relaxing and vasodilating hormones.The heart contains many neurohumoral receptors (alpha-1, beta-1, beta-2, beta-3, angiotensin II type 1 [AT1] and type 2 [AT2], muscarinic, endothelin, serotonin, adenosine, cytokine, natriuretic peptides); the roles of all of these receptors are not yet fully defined. In patients with heart failure, beta-1 receptors (which constitute 70% of cardiac beta receptors) are downregulated, probably in response to intense sympathetic activation. The result of downregulation is impaired myocyte contractility and increased heart rate.Plasma norepinephrine levels are increased, largely reflecting sympathetic nerve stimulation as plasma epinephrine levels are not increased. Detrimental effects include vasoconstriction with increased preload and afterload, direct myocardial damage including apoptosis, reduced renal blood flow, and activation of other neurohumoral systems, including the renin-angiotensin-aldosterone- vasopressin system.Vasopressin is released in response to a fall in BP via various neurohormonal stimuli. Increased vasopressin decreases renal excretion of free water, possibly contributing to hyponatremia in heart failure. Vasopressin levels in patients with HF and normal BP vary.Atrial natriuretic peptide is released in response to increased atrial volume and pressure; brain (B-type) natriuretic peptide (BNP) is released from the ventricle in response to ventricular stretching. These peptides enhance renal excretion of sodium, but in patients with HF, the effect is blunted by decreased renal perfusion pressure, receptor downregulation, and perhaps enhanced enzymatic degradation. In addition, elevated levels of natriuretic peptides exert a counter-regulatory effect on the renin-angiotensin-aldosterone system and catecholamine stimulation.Because endothelial dysfunction occurs in HF, fewer endogenous vasodilators (eg, nitric oxide, prostaglandins) are produced, and more endogenous vasoconstrictors (eg, endothelin) are produced, thus increasing afterload.The failing heart and other organs produce tumor necrosis factor (TNF) alpha. This cytokine increases catabolism and is possibly responsible for cardiac cachexia (loss of lean tissue ≥ 10%), which may accompany severely symptomatic HF, and for other detrimental changes. The failing heart also undergoes metabolic changes with increased free fatty acid utilization and decreased glucose utilization; these changes may become therapeutic targets.Changes with agingAge-related changes in the heart and cardiovascular system lower the threshold for expression of heart failure. Interstitial collagen within the myocardium increases, the myocardium stiffens, and myocardial relaxation is prolonged. These changes lead to a significant reduction in diastolic left ventricular function, even in healthy elderly people. Modest decline in systolic function also occurs with aging. An age-related decrease in myocardial and vascular responsiveness to beta-adrenergic stimulation further impairs the ability of the cardiovascular system to respond to increased work demands.As a result of these changes, peak exercise capacity decreases significantly (about 8%/decade after age 30), and CO at peak exercise decreases more modestly. This decline can be slowed by regular physical exercise. Thus, elderly patients are more prone than are younger ones to develop HF symptoms in response to the stress of systemic disorders or relatively modest cardiovascular insults. Stressors include infections (particularly pneumonia), hyperthyroidism, anemia, hypertension, myocardial ischemia, hypoxia, hyperthermia, renal failure, perioperative IV fluid loads, nonadherence to drug regimens or to low-salt diets, and use of certain drugs (particularly NSAIDs). |
|
|
|
What is farmers lung |
Symptoms Farmer's Lung is an allergic disease usually caused by breathing in the dust from moldy hay. However, dust from any moldy crop - straw, corn, silage, grain, or even tobacco - can also cause Farmer's Lung. ... "Pneumonitis" means inflammation of the lungs ("pneumon", Greek for lung). |
Hypersensitivity pneumonitis is a syndrome of cough, dyspnea, and fatigue caused by sensitization and subsequent hypersensitivity to environmental (frequently occupational) antigens. Acute, subacute, and chronic forms exist; all are characterized by acute interstitial inflammation and development of granulomas and fibrosis with long-term exposure. Diagnosis is based on a combination of history, physical examination, imaging tests, bronchoalveolar lavage, and biopsy. Short-term treatment is with corticosteroids; long-term treatment is antigen avoidance. Hypersensitivity pneumonitis is a syndrome of cough, dyspnea, and fatigue caused by sensitization and subsequent hypersensitivity to environmental (frequently occupational) antigens. Acute, subacute, and chronic forms exist; all are characterized by acute interstitial inflammation and development of granulomas and fibrosis with long-term exposure. Diagnosis is based on a combination of history, physical examination, imaging tests, bronchoalveolar lavage, and biopsy. Short-term treatment is with corticosteroids; long-term treatment is antigen avoidance. |
|
|
What is traction bronchiectasis |
Q |
|
|
|
Lewy body dementia Initial between Parkinson disease dementia and Alzheimer disease dementia |
Psychological aspects more VISUAL HALLUCINATIONS - COMPLEX DELUSIONS - BIZARRE
DELIRIUM LIKE FEATURES FLUCTUATING COGNITION ATTENTION AFFECTED MORE EARLY THAN MEMORY
EXTRAPYRAMIDAL SYMPTOMS EARLY THAN IN OTHER DEMENTIAS BUT DIFFERENT FROM THAT OF PARKINSON DISEASE ( SYMMETRICAL. TREMOR NOT SEEN EARLY)
Autonomic dysfunction common REM SLEEP BEHAVIOUR DISORDERS COMMON
Extreme sensitivity to antipsychotics is common |
|
|
|
What is methyldopa |
Methyldopa is in the alpha-2 adrenergic receptor agonist family of medication. It works by stimulating the brain to decrease the activity of the sympathetic nervous system. An alpha-2 adrenergic agonist that has both central and peripheral nervous system effects. Its primary clinical use is as an antihypertensive agent. [PubChem] |
|
|
|
How does survival after STEMI and NSTEMI changes with time |
In 6 months time and so on |
|
|
|
Heart failure causes |
Heart failure (HF) is a syndrome of ventricular dysfunction. Left ventricular failure causes shortness of breath and fatigue, and right ventricular failure causes peripheral and abdominal fluid accumulation; the ventricles can be involved together or separately.
Diagnosis is initially clinical, supported by chest x-ray, echocardiography, and levels of plasma natriuretic peptides.
Treatment includes patient education, diuretics, ACE inhibitors, angiotensin II receptor blockers, beta-blockers, aldosterone antagonists, neprilysin inhibitors, specialized implantable pacemakers/defibrillators and other devices, and correction of the cause(s) of the HF syndrome.
Etiology Both cardiac and systemic factors can impair cardiac performance and cause or aggravate heart failure
Causes of Heart Failure Cardiac Myocardial damage Myocardial infarction Myocarditis Cardiomyopathy Some chemotherapy drugs Valvular disorders Aortic stenosis Mitral regurgitation Arrhythmias Bradyarrhythmias Tachyarrhythmias
Conduction defects AV node block Left bundle branch block
Reduced substrate availability (eg, of free fatty acids or glucose) Ischemia
Infiltrative or matrix disorders Amyloidosis Chronic fibrosis (eg, systemic sclerosis) Hemochromatosis
Systemic Disorders that increase demand for CO AnemiaHyperthyroidismPaget disease
Disorders that increase resistance to output (afterload) Aortic stenosisHypertension
|
|
|
|
Heart failure classification |
Classification The most common classification of HF currently in use stratifies patients into
Heart failure with reduced ejection fraction ("systolic HF")Heart failure with preserved ejection fraction ("diastolic HF")
Heart failure with reduced ejection fraction (HFrEF) is defined as heart failure with LVEF ≤ 40%.
Heart failure with preserved ejection fraction (HFpEF) is defined as heart failure with LVEF ≥ 50%.
Patients with LVEF between 40% and 50% are in an intermediate zone, and have recently been categorized as HF with mid-range EF (HFmrEF) (1).The traditional distinction between left and right ventricular failure is somewhat misleading because the heart is an integrated pump, and changes in one chamber ultimately affect the whole heart. However, these terms indicate the major site of pathology leading to heart failure and can be useful for initial evaluation and treatment. Other common descriptive terms include acute or chronic; high output or low output; dilated or nondilated; and ischemic, hypertensive, or idiopathic dilated cardiomyopathy. Treatment differs based on whether the presentation is acute or chronic HF LV failure characteristically develops in ischemic heart disease, hypertension, mitral regurgitation, aortic regurgitation, aortic stenosis, most forms of cardiomyopathy, and congenital heart disorders (eg, ventricular septal defect, patent ductus arteriosus with large shunts). RV failure is most commonly caused by previous LV failure (which increases pulmonary venous pressure and leads to pulmonary hypertension, thus overloading the RV) or by a severe lung disorder (in which case it is called cor pulmonale). Other causes are multiple pulmonary emboli, RV infarction, pulmonary arterial hypertension, tricuspid regurgitation, tricuspid stenosis, mitral stenosis, pulmonary artery stenosis, pulmonic valve stenosis, pulmonary venous occlusive disease, arrhythmogenic RV cardiomyopathy, or congenital disorders such as Ebstein anomaly or Eisenmenger syndrome. Some conditions mimic RV failure, except cardiac function may be normal; they include volume overload and increased systemic venous pressure in polycythemia or overtransfusion, acute kidney injury with retention of sodium and water, obstruction of either vena cava, and hypoproteinemia due to any cause resulting in low plasma oncotic pressure and peripheral edema. Biventricular failure results from disorders that affect the whole myocardium (eg, viral myocarditis, amyloidosis, Chagas disease) or long-standing LV failure causing RV failure. High-output HF results from a persistently high cardiac output, which may eventually result in an inability of a normal heart to maintain adequate output. Conditions that may increase CO include severe anemia, end-stage liver disease, beriberi, thyrotoxicosis, advanced Paget disease, arteriovenous fistula, and persistent tachycardia. Cardiomyopathy is a general term reflecting disease of the myocardium. Most commonly, the term refers to a primary disorder of the ventricular myocardium that is not caused by congenital anatomic defects; valvular, systemic, or pulmonary vascular disorders; isolated pericardial, nodal, or conduction system disorders; or epicardial coronary artery disease (CAD). The term is sometimes used to reflect etiology (eg, ischemic vs hypertensive cardiomyopathy). Cardiomyopathy does not always lead to symptomatic HF. It is often idiopathic and is classified as dilated, congestive, hypertrophic, infiltrative-restrictive, or apical-ballooning cardiomyopathy (also known as takotsubo or stress cardiomyopathy). |
|
|
|
What are the symptoms and signs of heart failure |
Merck Manuals Professional EditionMerck ManualsFree - In Google PlayViewLogoMERCK MANUALProfessional VersionSystolic and diastolic heart failure HOMEMEDICAL TOPICSRESOURCESDRUG INFONEWSPROCEDURESQUIZZES & CASESABOUTLogoMERCK MANUALProfessional Version PROFESSIONAL / CARDIOVASCULAR DISORDERS / HEART FAILURE / HEART FAILURE (HF)Heart Failure (HF)(Congestive Heart Failure)By Sanjiv J. Shah, MD, Northwestern University Feinberg School of MedicineCLICK HERE FOR PATIENT EDUCATIONHeart failure (HF) is a syndrome of ventricular dysfunction. Left ventricular failure causes shortness of breath and fatigue, and right ventricular failure causes peripheral and abdominal fluid accumulation; the ventricles can be involved together or separately. Diagnosis is initially clinical, supported by chest x-ray, echocardiography, and levels of plasma natriuretic peptides. Treatment includes patient education, diuretics, ACE inhibitors, angiotensin II receptor blockers, beta-blockers, aldosterone antagonists, neprilysin inhibitors, specialized implantable pacemakers/defibrillators and other devices, and correction of the cause(s) of the HF syndrome.(For heart failure in children, see Overview of Congenital Cardiovascular Anomalies : Heart failure.)Heart failure affects about 6.5 million people in the US; > 960,000 new cases occur each year. About 26 million people are affected worldwide.PhysiologyPathophysiologyEtiologyClassificationSymptoms and SignsManifestations differ depending on the extent to which the LV and RV are initially affected. Clinical severity varies significantly and is usually classified according to the New York Heart Association (NYHA) system (see Table: New York Heart Association (NYHA) Classification of Heart Failure); the examples of ordinary activity may be modified for elderly, debilitated patients. Because HF has such a broad range of severity, some experts suggest subdividing NYHA class III into IIIA or IIIB. Class IIIB is typically reserved for those patients who recently had a heart failure exacerbation. The American College of Cardiology/American Heart Association has advocated a staging system for HF (A, B, C, or D) to highlight the need for HF prevention.A: High risk of HF but no structural or functional cardiac abnormalities or symptomsB: Structural or functional cardiac abnormalities but no symptoms of HFC: Structural heart disease with symptoms of HFD: Refractory HF requiring advanced therapies (eg, mechanical circulatory support, cardiac transplantation) or palliative careSevere LV failure may cause pulmonary edema or cardiogenic shock.TABLENew York Heart Association (NYHA) Classification of Heart FailureiconHistoryIn LV failure, the most common symptoms are dyspnea and fatigue due to increased pulmonary venous pressures, and low CO (at rest or inability to augment CO during exertion). Dyspnea usually occurs during exertion and is relieved by rest. As HF worsens, dyspnea can occur during rest and at night, sometimes causing nocturnal cough. Dyspnea occurring immediately or soon after lying flat and relieved promptly by sitting up (orthopnea) is common as HF advances. In paroxysmal nocturnal dyspnea (PND), dyspnea awakens patients several hours after they lie down and is relieved only after they sit up for 15 to 20 min. In severe HF, periodic cycling of breathing (Cheyne-Stokes respiration—a brief period of increased breathing [hyperpnea] followed by a brief period of no breathing [apnea])—can occur during the day or night; the sudden hyperpneic phase may awaken the patient from sleep. Cheyne-Stokes breathing differs from PND in that the hyperpneic phase is short, lasting only 10 to 15 sec, but the cycle recurs regularly, lasting 30 sec to 2 min. PND is associated with pulmonary congestion, and Cheyne-Stokes respiration with low CO. Sleep-related breathing disorders, such as sleep apnea, are common in HF and may aggravate HF. Severely reduced cerebral blood flow and hypoxemia can cause chronic irritability and impair mental performance.In RV failure, the most common symptoms are ankle swelling and fatigue. Sometimes patients feel a sensation of fullness in the abdomen or neck. Hepatic congestion can cause right upper quadrant abdominal discomfort, and stomach and intestinal congestion can cause early satiety, anorexia, and abdominal bloating.Less specific heart failure symptoms include cool peripheries, postural light-headedness, nocturia, and decreased daytime micturition. Skeletal muscle wasting can occur in severe biventricular failure and may reflect some disuse but also increased catabolism associated with increased cytokine production. Significant weight loss (cardiac cachexia) is an ominous sign associated with high mortality.In the elderly, presenting complaints may be atypical, such as confusion, delirium, falls, sudden functional decline, nocturnal urinary incontinence, or sleep disturbance. Coexisting cognitive impairment and depression may also influence assessment and therapeutic interventions and may be worsened by the HF.ExaminationGeneral examination may detect signs of systemic or cardiac disorders that cause or aggravate heart failure (eg, anemia, hyperthyroidism, alcoholism, hemochromatosis, atrial fibrillation with rapid rate, mitral regurgitation).In LV failure, tachycardia and tachypnea may occur. Patients with severe LV failure may appear visibly dyspneic or cyanotic, hypotensive, and confused or agitated because of hypoxia and poor cerebral perfusion. Some of these less specific symptoms (eg, confusion) are more common in the elderly.Central cyanosis (affecting all of the body, including warm areas such as the tongue and mucous membranes) reflects severe hypoxemia. Peripheral cyanosis of the lips, fingers, and toes reflects low blood flow with increased oxygen extraction. If vigorous massage improves nail bed color, cyanosis may be peripheral; increasing local blood flow does not improve color if cyanosis is central.Cardiac findings in HFrEF include a diffuse, sustained, and laterally displaced apical impulse; audible and occasionally palpable 3rd (S3) and 4th (S4) heart sounds, and an accentuated pulmonic component (P2) of the 2nd heart sound (S2). These abnormal heart sounds can also occur in HFpEF. A pansystolic murmur of mitral regurgitation at the apex may occur in either HFrEF or HFpEF. Pulmonary findings include early inspiratory basilar crackles that do not clear with coughing and, if pleural effusion is present, dullness to percussion and diminished breath sounds at the lung base(s).Signs of RV failure include nontender peripheral pitting edema (digital pressure leaves visible and palpable imprints, sometimes quite deep) in the feet and ankles; an enlarged and sometimes pulsatile liver palpable below the right costal margin; abdominal swelling and ascites; and visible elevation of the jugular venous pressure, sometimes with large a or v waves that are visible even when the patient is seated or standing (see Figure: Normal jugular vein waves.). Large V waves in the jugular veins are usually indicative of significant tricuspid regurgitation which is often present in RV failure. A paradoxical increase in the jugular venous pressure during inspiration (Kussmaul sign) is indicative of right-sided heart failure and can be seen in RV failure, restrictive cardiomyopathy, constrictive pericarditis, and severe tricuspid regurgitation. In severe cases of HF, peripheral edema can extend to the thighs or even the sacrum, scrotum, lower abdominal wall, and occasionally even higher. Severe edema in multiple areas is termed anasarca. Edema may be asymmetric if patients lie predominantly on one side.With hepatic congestion, the liver may be palpably enlarged or tender, and hepatojugular or abdominal-jugular reflux may be detected (see Neck veins). Precordial palpation may detect the left parasternal lift of RV enlargement, and auscultation may detect the murmur of tricuspid regurgitation or the RV S3 along the left sternal border; both findings are augmented upon inspiration. |
|
|
|
What is the prognosis of heart failure |
Merck Manuals Professional EditionMerck ManualsFree - In Google PlayViewLogoMERCK MANUALProfessional VersionSystolic and diastolic heart failure HOMEMEDICAL TOPICSRESOURCESDRUG INFONEWSPROCEDURESQUIZZES & CASESABOUTLogoMERCK MANUALProfessional Version PROFESSIONAL / CARDIOVASCULAR DISORDERS / HEART FAILURE / HEART FAILURE (HF)Heart Failure (HF)(Congestive Heart Failure)By Sanjiv J. Shah, MD, Northwestern University Feinberg School of MedicineCLICK HERE FOR PATIENT EDUCATIONHeart failure (HF) is a syndrome of ventricular dysfunction. Left ventricular failure causes shortness of breath and fatigue, and right ventricular failure causes peripheral and abdominal fluid accumulation; the ventricles can be involved together or separately. Diagnosis is initially clinical, supported by chest x-ray, echocardiography, and levels of plasma natriuretic peptides. Treatment includes patient education, diuretics, ACE inhibitors, angiotensin II receptor blockers, beta-blockers, aldosterone antagonists, neprilysin inhibitors, specialized implantable pacemakers/defibrillators and other devices, and correction of the cause(s) of the HF syndrome.(For heart failure in children, see Overview of Congenital Cardiovascular Anomalies : Heart failure.)Heart failure affects about 6.5 million people in the US; > 960,000 new cases occur each year. About 26 million people are affected worldwide.PhysiologyPathophysiologyEtiologyClassificationSymptoms and SignsDiagnosisPrognosisGenerally, patients with heart failure have a poor prognosis unless the cause is correctable. Five-year survival after hospitalization for heart failure is about 35% regardless of the patient's EF. In overt chronic HF, mortality depends on severity of symptoms and ventricular dysfunction and can range from 10 to 40%/yr.Specific factors that suggest a poor prognosis include hypotension, low ejection fraction, presence of CAD, troponin release, elevation of BUN, reduced GFR, hyponatremia, and poor functional capacity (eg, as tested by a 6-min walk test).BNP, NTproBNP, and risk scores such as the MAGGIC Risk Score and the Seattle Heart Failure model, are helpful to predict prognosis in HF patients as an overall group, although there is significant variation in survival among individual patients.HF usually involves gradual deterioration, interrupted by bouts of severe decompensation, and ultimately death, although the time course is being lengthened with modern therapies. However, death can also be sudden and unexpected, without prior worsening of symptoms.End-of-life careAll patients and family members should be taught about disease progression and the risk of sudden cardiac death. For some patients, improving quality of life is as important as increasing quantity of life. Thus, it is important to determine patients’ wishes about resuscitation (eg, endotracheal intubation, CPR) if their condition deteriorates, especially when HF is already severe.All patients should be reassured that symptoms will be relieved, and they should be encouraged to seek medical attention early if their symptoms change significantly. Involvement of pharmacists, nurses, social workers, and clergy (when desired), who may be part of an interdisciplinary team or disease management program already in place, is particularly important in end-of-life care. |
|
|
|
What are the causes for honey combing appearance in lungs |
EtiologyInterstitial lung diseases (ILD), also called diffuse parenchymal lung disease, is a broad classification encompassing mainly non-neoplastic and inflammatory lung diseases that cause alterations to the lung parenchyma in a diffuse pattern. Although a large and diverse list of ILDs have been described, the majority seen in clinical practice are idiopathic pulmonary fibrosis (IPF), chronic hypersensitivity pneumonitis (HP), collagen vascular disease (CVD)–associated ILD, and sarcoidosis. Idiopathic usual interstitial pneumonia (UIP) is the most common idiopathic interstitial pneumonia, and it is associated with a poor prognosis and eventual honeycomb changes.
Furthermore, nonspecific interstitial pneumonia (NSIP), although less common, can be associated with honeycomb changes.
CVDs include
systemic lupus erythematosus, rheumatoid arthritis, progressive systemic sclerosis (diffuse scleroderma), Sjögren syndrome, and dermatomyositis/polymyositis. An increased frequency of bronchiolar histotypes in lung carcinomas appears to be associated with IPF, in which abnormal bronchiolar proliferation occurs in transformed small airways in honeycomb lung regions. Dozens of drugs have been linked to ILD; however, methotrexate and bleomycin are the two agents most strongly associated with fibrosing interstitial pneumonia and they are capable of producing UIP and/or NSIP patterns.
Regardless of the underlying disease process, the universal pathophysiology is believed to be acute injury to lung parenchyma leading to chronic interstitial inflammation, tissue destruction, fibroblastic activation and proliferation, pulmonary fibrosis and, eventually, architectural remodeling with honeycomb changes. This process usually evolves over a period of months to years; however, it can be accelerated.
|
Can honey combing be seen in bronchial carcinoma |
|
|
Can beta blocker be given in acute heart failure |
Q |
|
|
|
Heart failure management acute and chronic |
Q |
|
|
|
What are the common precipitating factors of myxedema coma |
Myxedema coma is a physiologic decompensation of untreated hypothyroidism that is usually caused by a precipitating factor such as the following: Infection Exposure to cold temperatures Trauma Burns Cerebrovascular accident Myocardial infarction Congestive heart failure Respiratory acidosis Medications such as the following: Tranquilizers Sedatives Anesthetics Narcotics Amiodarone Rifampin Beta blockers Lithium Phenytoin Gastrointestinal hemorrhage Metabolic disturbances such as hypoglycemia, hyponatremia, acidosis, and hypercapnia Ingestion of raw bok choy |
|
|
|
Neurological manifestations of myxoedema |
Numerous complex regulatory mechanisms influence the development and function of the peripheral and central nervous system. Among them, hormones belong to the most potent regulatory factors. Various particles known for their hormonal activity serve as neurotransmitters. Additionally, hormones secreted systemically modulate the function of the nervous system both on the brain level and in peripheral organs. Thyroid function has been shown to play a crucial role in the proper cognitive development but also in many other aspects of nervous system activity, in mechanisms involving direct interaction with intrinsic regulatory circuits or indirectly by systemic effects exerted e.g. on the circulatory system or metabolic pathways. Due to these close relations with the nervous system function, disturbances of thyrometabolic state are associated with a vast spectrum of neurological signs and symptoms including: mood and cognitive disorders, headache, ophthalmoplegia, tremor and other movement disorders, muscle weakness etc. Both hyper- and hypothyroidism may cause psychiatric symptoms like depressive or anxiety disorder, memory deficits, executive inability and even psychosis. The severe decompensated hypothyroidism may result in myxoedema coma - a life-threatening condition with sequentially progressing encephalopathic symptoms. Steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAAT) represents another form of encephalopathic disorder associated with thyroid disease and causing potentially serious clinical complications. In the periphery, the thyrometabolic disturbances may affect muscle function resulting in subjective tiredness and low exercise tolerance and in some cases (especially in hypothyroidism) in objective myopathic signs. Also peripheral nervous system may be affected, mainly in hypothyroid patients, with greater tendency to develop peripheral polyneuropathy and entrapment neuropathies such as carpal tunnel or Guyone canal syndromes. Importantly, the autoimmune thyroid disease has been shown to coexist with other autoimmune processes which may potentially cause neurological symptoms such as myasthenia, Guillain-Barre syndrome or pernicious anaemia. Such conditions have to always be taken in consideration as differential diagnoses in patients presenting with neurological signs and symptoms associated with thyroid disease. |
Hypothyroidism can affect the central and peripheral nervous systems at multiple levels, producing a diverse array of neurologic symptoms and signs. Clinicians should be particularly aware of the diagnostic and management issues related to myxedema coma, dementia, myopathy, and polyneuropathy. Carpal tunnel syndrome may be the most common neurologic abnormality associated with hypothyroidism. The importance of hypothyroidism as a “reversible cause of dementia” remains unclear. The diverse manifestations of Hashimoto encephalopathy, a syndrome that appears to be inflammatory rather than a direct result of inadequate thyroid hormone levels, are important to recognize because the syndrome typically responds to high-dose steroid treatment .Key points • Hypothyroidism can affect practically every level of the central and peripheral nervous system. • Although coma due to severe hypothyroidism (myxedema coma) is rare, it should always be considered in comatose patients without a clear cause because it requires rapid and specific treatment. • Carpal tunnel syndrome is the most common peripheral nerve manifestation of hypothyroidism. • An elevated thyrotropin level is the key diagnostic finding in primary hypothyroidism, which is the most common form of hypothyroidism. Historical note and terminology Hypothyroidism is the clinical condition that results from inadequate synthesis of thyroid hormone. |
|
|
Significance of prolonged PR interval within 24 h of MI |
DiscussionThis is our first recognized study to show that PR prolongation alone is a strong and independent predictor for CV death, new-onset ischemic stroke and MI, and combined CV endpoints including CHF among patients with CAD or risk equivalent. We also assessed the vascular phenotype of these high-risk CV patients and found that PR prolongation was independently related to increased carotid IMT. Interestingly, despite the stated observations, adjustment for carotid IMT did not materially alter the prediction of PR prolongation for incident CV events, which suggests that atherosclerosis alone does not fully explain the pathophysiological mechanism along the pathway of PR prolongation leading to clinical events.The clinical implications of this study are multi-fold. Firstly, as opposed to the traditional school of thought that considered PR prolongation as a benign ECG feature [1], this study robustly showed that PR prolongation is strongly predictive of a collective plethora of pathophysiologically related CV outcomes, including MI, ischemic stroke, CHF and CV death. Recent epidemiological studies (ARIC (2) and Framingham [3]) found higher risk of atrial fibrillation associated with PR prolongation. Our findings suggest that the pathophysiological implications of PR prolongation is far more than atrial fibrillation, and entails MI, ischemic stroke and CHF, all of which could be commonly characterized by impaired systemic vascular function [7–9]. PR prolongation alone, regardless of other pertinent CV risk factors [12] such as those delineated in the CHADS2 scores [5], is a simple and independent CV risk predictor. Secondly, the magnitude of excess risk of PR prolongation was substantial for various adverse outcomes consistently (new-onset ischemic stroke: 4-fold excess risk; new-onset MI, 7-fold excess risk; CV death: 15-fold excess risk), thus reflecting PR prolongation has strong utility for CV prediction among high-risk patients. Thirdly, while the recent Heart and Soul Study [4] showed that PR prolongation predicted heart failure hospitalization and CV death amongst patients with stable CAD, our study further showed that PR prolongation has strong predictive applicability for CV events in a much wider spectrum of high-risk patients with prior CAD or other risk equivalents, including diabetes and stroke. Fourthly, the escalated risk of new-onset MI emerged at a lower PR interval cut-off of >162 ms, consistent with our prior exploratory study in terms of subclinical changes in endothelial dysfunction [6], and calls into question that early pathological changes may well precede the conventional cut-off of PR prolongation at >200 ms.Mechanisms underlying our clinical observation are likely multi-dimensional. Firstly, PR prolongation representing delayed atrioventricular conduction could be an ECG manifestation of CV ageing [13]. Atherosclerosis is increasingly recognized as a multi-factorial degenerative disease that is closely related to the aging process. Our analysis indicated that PR prolongation could be an excellent clinical marker of CV ageing since adjustment for chronological age did not obliterate the prediction of carotid IMT and CV events by PR prolongation. Secondly, myocardial fibrosis may involve the cardiac conduction system and is closely linked to the presence of CV risk factors including CAD [14], hypertension [15], diabetes [16], hyperlipidemia [17] and inflammation [18]. Thus PR prolongation may be a risk marker that reflects the clustering of these important risk factors. Nevertheless, PR prolongation may also reflect higher underlying vagal tone [19], especially in the young [1], other than degenerative damage of the cardiac conduction system which predominantly affects the elderly [13]. In our study, comprehensive adjustment for important CV risk factors, even when including widened QRS complex signifying overt ventricular electrical dyssynchrony or widespread conduction system disease, did not abolish PR prolongation as an independent CV predictor. Therefore a third potential explanation arises: that PR prolongation may be causal to adverse CV events. Indeed, PR prolongation, be it vagal- or degenerative-predominant, may mediate its adverse vascular effects via increased intra-atrial pressure consequential to slowed atrioventricular conduction, resulting in neurohormonal activation [20]. Adverse neurohormonal changes including raised aldosterone may be reversible on sinus rhythm reversion [21]. Aldosterone is associated with a constellation of pro-atherosclerotic changes including raised inflammatory response [22], reduced circulating endothelial progenitor cell quantity and function [23] resulting in increased arterial stiffness [24]. Importantly, mineralocorticoid receptor blockade has been shown clinically to improve atherosclerotic changes [25]. Indeed, if neurohormonal activation is the explanatory mechanism that underlies PR prolongation-related vascular dysfunction, such findings may translate into potentially important preventive implications through pharmacological modulation of the neurohormonal system.Vascular function as indicated by carotid IMT is a strong, independent and consistent predictor of both incident ischemic stroke and CV events overall among healthy subjects from diverse ethnicities [26]. Increased carotid IMT also predicts elevated risks of CV complications in patients with diabetes [27], hypertension [28] and prior atherosclerotic diseases [29]. In this study, the magnitude of increased carotid IMT by +0.07 mm accounted for by PR prolongation is clinically significant, since each 0.1 mm increase of carotid IMT could translate into 18% excess risk of recurrent CV events [30]. On top of their greater susceptibility to CV events in general, patients with prior CAD also have increased risk of incident ischemic stroke, with diabetes being an additional independent predictor [31, 32]. Our study further suggests that the adverse prognostic value of PR prolongation is independent and over that of carotid IMT for CV events in high-risk patients. Whether impaired vascular function is indeed along the pathophysiological pathway or simply a bystander will require further mechanistic studies. Future studies could be strengthened by incorporating multiple vascular assessment modalities to ensure consistent findings and scrutinise further mechanisitc insights.It should be noted that from our study, the magnitude of risk associations of PR interval prolongation with new-onset CV events was striking, and that IMT did not fully explain such associations, suggesting that alternative mediating pathophysiological mechanisms could be present. Also, the HR estimates are relatively large. Despite our meticulous efforts in the study design and data analysis to carefully minimize biases or residual confounding, potential risk of bias and confounding cannot be excluded totally. Findings from the exploratory endpoint analyses should also best be verified in another independent study. Furthermore, the study of incident atrial fibrillation was not included in the current study, which should be explored in the future.ConclusionsIn this prospective study of patients with CAD or risk-equivalent, PR prolongation strongly and independently predicts CV death, new-onset MI, ischemic stroke, and combined CV endpoints including CHF. Increased risk of MI emerged at a cut-off as low as PR interval > 162 ms. Whether adverse vascular function in PR prolongation is an intermediate phenotype or represent a mediating pathway prior to clinical events warrants further investigations. |
An extremely prolonged PR interval may occur in First Degree Atrioventricular Block and it may be associated with Atrioventricular Dissociation and Pseudo-Pacemaker Syndrome which may pose diagnostic and management challenges. This suggests that not all cases of First Degree Atrioventricular Block are benign and so should be sub-classified based on degree of PR interval prolongation and associated electrical abnormalities. |
|
|
Anterior mi manifestations |
This site is intended for healthcare professionalsMedscape LogoDrugs & Diseases > CardiologyComplications of Myocardial InfarctionUpdated: Dec 18, 2014 Author: Ashok K Kondur, MD; Chief Editor: Eric H Yang, MD more...Share FeedbackSECTIONSOverviewMyocardial infarction (MI) due to coronary artery disease is a leading cause of death in the United States, where more than 1 million people have acute myocardial infarctions (AMIs) each year. [1]The advent of coronary care units and early reperfusion therapy (lytic or percutaneous coronary intervention) has substantially decreased in-hospital mortality rates and has improved the outcome in survivors of the acute phase of MI.Complications of MI include arrhythmic, mechanical, and inflammatory (early pericarditis and post-MI syndrome) sequelae, as well as left ventricular mural thrombus (LVMT). In addition to these broad categories, right ventricular (RV) infarction and cardiogenic shock are other possible complications of acute MI. (See the image below.)Modified 2-dimensional (top) echocardiogram and coModified 2-dimensional (top) echocardiogram and color flow Doppler image (bottom). Apical 4-chamber views show a breach in the interventricular septum and free communication between ventricles through a large apical septum ventricular septal defect in a patient who recently had an anterior myocardial infarction.View Media GalleryFor other discussions on myocardial infarction, see Myocardial Infarction, Right Ventricular Infarction, Imaging of Acute Myocardial Infarcts, and Use of Cardiac Markers in the Emergency Department.Arrhythmic Complications of MIAbout 90% of patients who have an acute myocardial infarction (AMI) develop some form of cardiac arrhythmia during or immediately after the event. In 25% of patients, such rhythm abnormalities manifest within the first 24 hours. In this group of patients, the risk of serious arrhythmias, such as ventricular fibrillation, is greatest in the first hour and declines thereafter. The incidence of arrhythmia is higher with an ST-elevation myocardial infarction (STEMI) and lower with a non–ST-elevation myocardial infarction (NSTEMI). [2]The clinician must be aware of these arrhythmias, in addition to reperfusion strategies, and must treat those that require intervention to avoid exacerbation of ischemia and subsequent hemodynamic compromise. Most peri-infarct arrhythmias are benign and self-limited. However, those that result in hypotension, increase myocardial oxygen requirements, and/or predispose the patient to develop additional malignant ventricular arrhythmias should be aggressively monitored and treated.Pathophysiology of arrhythmic complicationsAMI is characterized by generalized autonomic dysfunction that results in enhanced automaticity of the myocardium and conduction system. Electrolyte imbalances (eg, hypokalemia and hypomagnesemia) and hypoxia further contribute to the development of cardiac arrhythmia. The damaged myocardium acts as substrate for re-entrant circuits, due to changes in tissue refractoriness.Enhanced efferent sympathetic activity, increased concentrations of circulating catecholamines, and local release of catecholamines from nerve endings in the heart muscle itself have been proposed to play roles in the development of peri-infarction arrhythmias. Furthermore, transmural infarction can interrupt afferent and efferent limbs of the sympathetic nervous system that innervates myocardium distal to the area of infarction. The net result of this autonomic imbalance is the promotion of arrhythmias.Classification of peri-infarction arrhythmiasPeri-infarction arrhythmias can be broadly classified into the following categories:Supraventricular tachyarrhythmias, including sinus tachycardia, premature atrial contractions, paroxysmal supraventricular tachycardia, atrial flutter, and atrial fibrillationAccelerated junctional rhythmsBradyarrhythmias, including sinus bradycardia and junctional bradycardiaAtrioventricular (AV) blocks, including first-degree AV block, second-degree AV block, and third-degree AV blockIntraventricular blocks, including left anterior fascicular block, right bundle branch block (RBBB), and left bundle branch block (LBBB)Ventricular arrhythmias, including premature ventricular contractions (PVCs), accelerated idioventricular rhythm, ventricular tachycardia, and ventricular fibrillationReperfusion arrhythmiasArrhythmic Complications: Supraventricular TachyarrhythmiasSinus tachycardia is associated with enhanced sympathetic activity and can result in transient hypertension or hypotension. The elevated heart rate increases myocardial oxygen demand, and a decreased length of diastole compromises coronary flow, worsening myocardial ischemia.Causes of persistent sinus tachycardia include the following:PainAnxietyHeart failureHypovolemiaHypoxiaAnemiaPericarditisPulmonary embolismIn the setting of an AMI, sinus tachycardia must be identified, and appropriate treatment strategies must be devised. Treatment strategies include adequate pain medication, diuresis to manage heart failure, oxygenation, volume repletion for hypovolemia, administration of anti-inflammatory agents to treat pericarditis, and use of beta-blockers and/or nitroglycerin to relieve ischemia.Premature atrial contractionsPremature atrial contractions often occur before the development of paroxysmal supraventricular tachycardia, atrial flutter, or atrial fibrillation. The usual cause of these extra impulses is atrial distention due to increased left ventricular (LV) diastolic pressure or inflammation associated with pericarditis.No specific therapy is indicated. However, attention should be given to identifying the underlying disease process, particularly occult heart failure.Paroxysmal supraventricular tachycardiaThe incidence of a paroxysmal supraventricular tachycardia in the setting of an AMI is less than 10%. In the absence of definitive data in the patient with AMI, the consensus is that adenosine can be used when hypotension is not present. In patients without clinically significant LV failure, intravenous diltiazem or a beta-blocker can be used instead. In patients who develop severe heart failure or hypotension, synchronized electrical cardioversion is required.Atrial flutterAtrial flutter occurs in less than 5% of patients with AMI. Atrial flutter is usually transient and results from sympathetic overstimulation of the atria.Treatment strategies for persistent atrial flutter are similar to those for atrial fibrillation, except that ventricular-rate control with drugs is less easily accomplished with atrial flutter than with atrial fibrillation. Therefore, synchronized electrical cardioversion (beginning with 50 J, or the biphasic equivalent) may be needed relatively promptly because of a decrease coronary blood flow and/or hemodynamic compromise. For patients whose atrial flutter is refractory to medical therapy, overdrive atrial pacing may be considered.Atrial fibrillationThe rate of atrial fibrillation is 10-15% among patients who have AMIs. The onset of atrial fibrillation in the first hours of AMI is usually caused by LV failure, ischemic injury to the atria, or RV infarction. Pericarditis and all conditions leading to elevated left atrial pressure can also lead to atrial fibrillation in association with an AMI. The presence of atrial fibrillation during an AMI is associated with an increased risk of mortality and stroke, particularly in patients who have anterior-wall MIs.Immediate electrical cardioversion is indicated for the patient in unstable condition, such as one with new or worsening ischemic pain and/or hypotension. Synchronized electrical cardioversion to treat atrial fibrillation begins with 200 J (or the biphasic equivalent). Conscious sedation (preferred) or general anesthesia is advisable prior to cardioversion.For patients in stable condition, controlling the ventricular response is the immediate objective. If the atrial fibrillation does not respond to cardioversion, IV amiodarone [3] or IV digoxin (in patients with LV dysfunction or heart failure) can be used to achieve ventricular rate control.For patients who do not develop hypotension, a beta-blocker can be used. For example, metoprolol may be given in 5-mg intravenous boluses every 5-10 min, with a maximum dose of 15 mg. Intravenous diltiazem is an alternative for slowing the ventricular rate, but it should be used with caution in patients with moderate-to-severe heart failure. In patients with new-onset sustained tachycardia (absent before MI), conversion to sinus rhythm should be considered as an option.Atrial fibrillation and atrial flutter confer an increased risk of thromboembolism (see Deep Venous Thrombosis and Pulmonary Embolism). Therefore, anticoagulation with either unfractionated heparin or low molecular weight heparin (LMWH) should be started if contraindications are absent. It is unclear whether anticoagulation is needed in cases of transient atrial fibrillation and how long after the onset of atrial fibrillation should the anticoagulation be started.Arrhythmic Complications: Accelerated Junctional RhythmAn accelerated junctional rhythm results from increased automaticity of the junctional tissue that leads to a heart rate of 70-130 bpm. This type of dysrhythmia is most common in patients who develop inferior myocardial infarctions. Treatment is directed at correcting the underlying ischemia.Arrhythmic Complications: BradyarrhythmiasSinus bradycardiaSinus bradycardia is a common arrhythmia in patients with inferior or posterior acute myocardial infarctions (AMIs). The highest incidence, 40%, is observed in the first 1-2 hours after AMI.The likely mechanism leading to bradycardia and hypotension is stimulation of cardiac vagal afferent receptors that result in efferent cholinergic stimulation of the heart. In the early phases of an AMI, resultant sinus bradycardia may actually be protective, reducing myocardial oxygen demand. Clinically significant bradycardia that decreases cardiac output and hypotension may result in ventricular arrhythmias and should, therefore, be treated aggressively. Isolated sinus bradycardia is not associated with an increase in the acute mortality risk, and therapy is typically unnecessary when the patient has no adverse signs or symptoms.When emergency therapy is indicated (eg, in a patient with a sinus rate of < 40 bpm with hypotension), atropine sulfate 0.5-1 mg may be given every 3-5 minutes to a maximum of 0.03-0.04 mg/kg. The inability to reverse hypotension with atropine in patients who develop sinus bradycardia and inferior MI suggests volume depletion and/or RV infarction.When atropine is ineffective and the patient is symptomatic or hypotensive, transcutaneous or transvenous pacing is indicated (see our main article on External Pacemakers). Denervate, transplanted hearts do not respond to atropine and, therefore, require cardiac pacing.If these interventions fail, additional pharmacologic intervention may be useful. Examples are dopamine 5-20 mcg/kg/min given intravenously, epinephrine 2-10 mcg/min, and/or dobutamine.Junctional bradycardiaJunctional bradycardia is a protective AV junctional escape rhythm at a rate of 35-60 bpm in patients who have an inferior MI. This arrhythmia is not usually associated with hemodynamic compromise, and treatment is typically not required.Arrhythmic Complications: AV and Intraventricular BlocksFirst-degree AV blockFirst-degree AV block is characterized by prolongation of the PR interval to longer than 0.20 seconds. This type of block occurs in approximately 15% of patients who have an acute myocardial infarction (AMI), most commonly an inferior infarction. Almost all patients who develop first-degree AV block have conduction disturbances above the His bundle. In these patients, the progression to complete heart block or ventricular asystole is rare. No specific therapy is indicated unless associated hemodynamic compromise is present.Calcium channel blockers and beta-blockers may cause or exacerbate a first-degree AV block, but they should be stopped only if hemodynamic impairment or a higher-degree block occurs. For a first-degree AV block associated with sinus bradycardia and hypotension, atropine should be administered. Continued cardiac monitoring is advisable in view of possible progression to higher degrees of block.Second-degree AV blockMobitz type I, or Wenckebach, AV block occurs in approximately 10% of patients who have an AMI and accounts for 90% of all patients who have an AMI and a second-degree AV block. A second-degree AV block is associated with a narrow QRS complex and is most commonly associated with an inferior MI. It does not affect the patient's overall prognosis.A Mobitz type I block does not necessarily require treatment. If the heart rate is inadequate for perfusion, immediate treatment with atropine 0.5-1 mg administered intravenously is indicated. Transcutaneous or temporary transvenous pacing is rarely required.A Mobitz type II AV block accounts for 10% of all second-degree AV blocks (overall rate of < 1% in the setting of AMI). A Mobitz type II block is characterized by a wide QRS complex, and it is almost always associated with anterior infarction. This type of block often progresses suddenly to a complete heart block.Mobitz type II AV blocks are associated with a poor prognosis, as the mortality rate associated with their progression to a complete heart block is approximately 80%. Therefore, this type of second-degree AV block should be immediately treated with transcutaneous pacing or atropine. Atropine helps in about 50% of cases, but it occasionally worsens the block with an increased heart rate. A temporary transvenous pacemaker, and possibly a permanent demand pacemaker, must ultimately be placed.Third-degree AV blockA third-degree AV block (ie, a complete heart block), occurs in 5-15% of patients who have an AMI and may occur with anterior or inferior infarctions. In patients with inferior infarctions, this type of block usually develops gradually, progressing from first-degree or a type I second-degree block. In most patients, the level of the block is supranodal or intranodal, and the escape rhythm is usually stable with a narrow QRS and rates exceeding 40 bpm. In 30% of patients, the block is below the His bundle, where it results in an escape rhythm with a rate slower than 40 bpm and a wide QRS complex.Complete heart block in patients with an inferior MI usually responds to atropine. In most patients, it resolves within a few days without the need for a temporary or permanent pacemaker. The mortality rate for patients with inferior MI who develop complete heart block is approximately 15% unless a coexisting RV infarction is present, in which case the mortality rate is higher.Immediate treatment with atropine is indicated for patients with third-degree AV blocks. As with therapy for a Mobitz type II block, this treatment may not help and may sometimes worsen the block. Temporary transcutaneous or transvenous pacing is indicated for symptomatic patients whose condition is unresponsive to atropine. Permanent pacing should be considered in patients with persistent symptomatic bradycardia that remains unresolved with lysis or percutaneous coronary intervention.In patients with an anterior MI, an intraventricular block or a Mobitz type II AV block usually precedes a third-degree AV block. The third-degree block occurs suddenly and is associated with a high mortality rate. The Cardiac Arrhythmias and Risk Stratification After Myocardial Infarction (CARISMA) trial monitored patients with acute myocardial infarction and reduced left ventricular ejection fraction and found that high-degree atrioventricular block was the most powerful predictor of cardiac death. [4] Patients with these blocks typically have unstable escape rhythms with wide QRS complexes and at rates of less than 40 bpm.Immediate treatment with atropine and/or transcutaneous pacing is indicated. This is followed by temporary transvenous pacing. Patients with an anterior MI who develop a third-degree AV block and who survive to hospitalization often receive a permanent pacemaker.Intraventricular blocksConduction from the His bundle is transmitted through 3 fascicles: the anterior division of the left bundle, the posterior division of the left bundle, and the right bundle. An abnormality of electrical conduction in 1 or more of these fascicles is noted in about 15% of patients with AMI. Isolated left anterior fascicular block (LAFB) occurs in 3-5% of patients with AMI; progression to complete AV block is uncommon. Isolated left posterior fascicular block occurs in only 1-2% of patients who have an AMI. The blood supply of the posterior fascicle is larger than that of the anterior fascicle; therefore, a block here is associated with a relatively large infarct and high mortality rate.The right bundle branch receives its dominant blood supply from the left anterior descending (LAD) artery. Therefore, a new RBBB, which is seen in approximately 2% of patients with AMI, suggests a large infarct territory. However, progression to complete heart block is uncommon. In patients who develop an anterior MI and a new RBBB, the substantial risk for death is mostly from cardiogenic shock, which is presumably due to the large size of the myocardial infarct.The combination of RBBB with an LAFB is known as bifascicular block and commonly occurs with occlusion of the proximal LAD coronary artery. The risk of developing complete AV block is heightened, but complete block is still uncommon. Mortality is mostly related to the amount of muscle loss. Bifascicular block in the presence of first-degree AV block is called a trifascicular block. In 40% of patients, a trifascicular block progresses to a complete heart block.Arrhythmic Complications: Ventricular ArrhythmiasPremature ventricular contractionsIn the past, frequent premature ventricular contractions (PVCs) were considered to represent warning arrhythmias and indicators of impending malignant ventricular arrhythmias. However, presumed warning arrhythmias are frequently observed in patients who have an acute myocardial infarction (AMI) and who never develop ventricular fibrillation. On the converse, primary ventricular fibrillation often occurs without antecedent premature ventricular ectopy.For these reasons, prophylactic suppression of PVCs with antiarrhythmic drugs, such as lidocaine, is no longer recommended. Prophylaxis has been associated with an increased risk of fatal bradycardia or asystole because of the suppression of escape pacemakers.Given this evidence, most clinicians pursue a conservative course when PVCs are observed in a patient with an AMI, and they do not routinely administer prophylactic antiarrhythmics. Instead, attention should be directed toward correcting any electrolytic or metabolic abnormalities, plus identifying and treating recurrent ischemia.Accelerated idioventricular rhythmAn accelerated idioventricular rhythm is seen in as many as 20% of patients who have an AMI. This pattern is defined as a ventricular rhythm characterized by a wide QRS complex with a regular escape rate faster than the atrial rate, but less than 100 bpm. AV dissociation is frequent. Slow, nonconducted P waves are seen; these are unrelated to the fast, wide QRS rhythm.Most episodes are short and terminate spontaneously. They occur with equal frequency in anterior and inferior infarctions. The mechanism might involve (1) the sinoatrial node or the AV node, which may sustain structural damage and depress nodal automaticity, and/or (2) an abnormal ectopic focus in the ventricle that takes over as the dominant pacemaker.The presence of accelerated idioventricular rhythm does not affect the patient's prognosis; no definitive evidence has shown that an untreated occurrence increases the incidence of ventricular fibrillation or death. This rhythm occurs somewhat more frequently in patients who develop early reperfusion than in others; however, it is neither sensitive nor specific as a marker of reperfusion.Temporary pacing is not indicated unless the rhythm is sustained and results in hypotension or ischemic symptoms. An accelerated idioventricular rhythm represents an appropriate escape rhythm. Suppression of this escape rhythm with an antiarrhythmic drug can result in clinically significant bradycardia or asystole. Therefore, an accelerated idioventricular rhythm should be left untreated.Nonsustained ventricular tachycardiaNonsustained ventricular tachycardia is defined as 3 or more consecutive ventricular ectopic beats at a rate of greater than 100 bpm and lasting less than 30 seconds. In patients who experience multiple runs of nonsustained ventricular tachycardia, the risk for sudden hemodynamic collapse may be substantial.Nonetheless, nonsustained ventricular tachycardia in the immediate peri-infarction period does not appear to be associated with an increased mortality risk, and no evidence suggests that antiarrhythmic treatment offers a morbidity or mortality benefit. However, nonsustained ventricular tachycardia occurring more than 48 hours after infarction in patients with LV systolic dysfunction (LV ejection fraction < 0.40) poses an increased risk for sudden cardiac death; electrophysiologic testing and appropriate therapy are indicated in these patients.Multiple episodes of nonsustained ventricular tachycardia require intensified monitoring and attention to electrolyte imbalances. Serum potassium levels should be maintained above 4.5 mEq/L, and serum magnesium levels should be kept above 2.0 mEq/L. Ongoing ischemia should aggressively be sought and corrected if found.Sustained ventricular tachycardiaSustained ventricular tachycardia is defined as 3 or more consecutive ventricular ectopic beats at a rate greater than 100 bpm and lasting longer than 30 seconds or causing hemodynamic compromise that requires intervention. Monomorphic ventricular tachycardia is most likely to be caused by a myocardial scar, whereas polymorphic ventricular tachycardia may be most responsive to measures directed against ischemia. Sustained polymorphic ventricular tachycardia after an AMI is associated with a hospital mortality rate of 20%.Emergency treatment of sustained ventricular tachycardia is mandatory because of its hemodynamic effects and because it frequently deteriorates into ventricular fibrillation. Rapid polymorphic ventricular tachycardia (rate >150 bpm) associated with hemodynamic instability should be treated with immediate direct-current unsynchronized cardioversion of 200 J (or biphasic energy equivalent). Monomorphic ventricular tachycardia should be treated with a synchronized discharge of 100 J (or biphasic energy equivalent).If sustained ventricular tachycardia is well tolerated, antiarrhythmic therapy with amiodarone (drug of choice) or procainamide may be attempted before electrical cardioversion. Precipitating causes, such as electrolyte abnormalities, acid-base disturbances, hypoxia, or medication, should be sought and corrected. For persistent or recurrent ventricular tachycardia, overdrive pacing may be effective in electrically converting the patient's rhythm to a sinus rhythm.Ventricular fibrillationThe incidence of primary ventricular fibrillation is greatest in the first hour after the onset of infarct (4.5%) and declines rapidly thereafter. Approximately 60% of episodes occur within 4 hours, and 80% occur within 12 hours.Secondary or late ventricular fibrillation occurring more than 48 hours after an MI is usually associated with pump failure and cardiogenic shock. Factors associated with an increased risk of secondary ventricular fibrillation are a large infarct, an intraventricular conduction delay, and an anteroseptal AMI. Secondary ventricular fibrillation in conjunction with cardiogenic shock is associated with an in-hospital mortality rate of 40-60%.Treatment for ventricular fibrillation is unsynchronized electrical countershock with at least 200-300 J (or biphasic energy equivalent) administered as rapidly as possible. Each minute after the onset of uncorrected ventricular fibrillation is associated a 10% decrease in the likelihood of survival. Restoration of synchronous cardiac electrical activity without the return of effective contraction (ie, electromechanical dissociation, or pulseless electrical activity) is generally due to extensive myocardial ischemia and/or necrosis or cardiac rupture.Antiarrhythmics, such as intravenous amiodarone and lidocaine, facilitate successful electrical defibrillation and help prevent recurrent or refractory episodes. After ventricular fibrillation is successfully converted, antiarrhythmic therapy is generally continued as a constant intravenous infusion for 12-24 hours.Prophylactic lidocaine reduces the incidence of ventricular fibrillation, but it is not used because it seems to be associated with an excessive mortality risk owing to bradycardic and asystolic events. [5] On the other hand, early use of beta-blockers in patients with AMI reduces the incidence of ventricular fibrillation as well as death. [6]Arrhythmic Complications: Reperfusion ArrhythmiasIn the past, the sudden onset of rhythm disturbances after thrombolytic therapy in patients with AMI was believed to be a marker of successful coronary reperfusion. However, a high incidence of identical rhythm disturbances is observed in patients with AMI in whom coronary reperfusion is unsuccessful. Therefore, these so-called reperfusion arrhythmias are neither sensitive nor specific for reperfusion and should be treated as discussed under Accelerated Idioventricular Rhythm in the Arrhythmic Complications: Ventricular Arrhythmias section above.Mechanical Complications of MIThe 3 major mechanical complications of AMI are ventricular free wall rupture (VFWR), ventricular septal rupture (VSR), and papillary muscle rupture with severe mitral regurgitation (MR). Each of these complications can result in cardiogenic shock. Clinical issues related to these mechanical problems are discussed below. (See also Myocardial Rupture.)Overview of ventricular free wall ruptureVFWR is the most serious complication of AMI. VFWR is usually associated with large transmural infarctions and antecedent infarct expansion. It is the most common cause of death, second only to LV failure, and it accounts for 15-30% of the deaths associated with AMI. Incontrovertibly the most catastrophic of mechanical complications, VFWR leads to acute hemopericardium and death from cardiac tamponade.The overall incidence of VFWR ranges from 0.8-6.2%. The incidence of this complication has declined over the years with better 24 hour systolic blood pressure control; increased use of reperfusion therapy, beta blockers, and ACE inhibitors; and decreased use of heparin [7] .Data from the National Registry of Myocardial Infarction (NRMI) showed an elevated incidence of in-hospital mortality among patients who received thrombolytic therapy (12.1%) than among patients who did not (6.1%). [8] In the Thrombolysis in Myocardial Infarction Phase II (TIMI II) trial, 16% of patients died from cardiac rupture within 18 hours of therapy. [9] Patients who underwent percutaneous transluminal coronary angioplasty (PTCA) had an incidence of free wall rupture lower than that of patients receiving thrombolytic therapy.Risk factors for VFWR include advanced age greater than 70 years, female sex, no previous MIs, Q waves on ECG, hypertension during the initial phase of STEMI, corticosteroid or NSAID use, and fibrinolytic therapy more than 14 hours after STEMI onset. Patients with a history of angina pectoris, previous AMI, multivessel coronary disease, and chronic heart failure are less likely than others to develop VFWR of the LV because they develop collaterals and ischemic preconditioning. [8, 10, 11]Clinical presentation of VFWRVFWRs are dramatic; they present acutely or occasionally subacutely as pseudoaneurysms; and they most often involve the anterior or lateral wall of the LV. Most VFWRs occur within the first week after AMI.Becker et al classified the following 3 types of VFWRs [12] :Type I - an abrupt slitlike tear that is frequently associated with anterior infarcts and that occurs early (within 24 h)Type II - an erosion of infarcted myocardium at the border between the infarcted and viable myocardiumType III - an early aneurysm formation correlated with older and severely expanded infarctsType III usually occurs later than type I or type II ruptures. Thrombolytic therapy accelerates the occurrence of cardiac rupture in Becker type I and type II VFWRs. In severely expanded infarctions (type III), thrombolytic therapy decreases the incidence of cardiac rupture.A pseudoaneurysm is formed when adjacent pericardium and hematoma seals off a myocardial rupture or perforation. The wall of a pseudoaneurysm is most often visualized as an aneurysmal outpouching that communicates with the LV cavity by means of a narrow neck. This wall is composed of pericardium and organized thrombus and/or hematoma. It is devoid of myocardial elements, whereas a true aneurysm has all the elements of the original myocardial wall and a relatively wide base. The pseudoaneurysm may vary in size and is at high risk of rupturing.Clinical presentations of VFWR vary depending on the acuity, location, and size of the rupture. Patients with acute VFWR present with severe chest pain, abrupt electromechanical dissociation or asystole, hemodynamic collapse, and possibly death. In about one third of the patients, the course is subacute, and they present with symptoms such as syncope, hypotension, shock, arrhythmia, and prolonged and recurrent chest pain.Diagnosis of VFWREarly diagnosis of VFWRs and intervention are critical to patient survival. A high index of suspicion is required when patients with AMI present with severe chest pain, shock or arrhythmias, and abrupt development of electromechanical dissociation. ECG signs of impending VFWR have limited specificity but include sinus tachycardia, intraventricular conduction defect, and persistent or recurrent ST-segment elevation.Echocardiography is the diagnostic tool of choice. The key diagnostic finding is a moderate-to-large pericardial effusion with clinical and echocardiographic signs of impending pericardial tamponade. In patients with cardiac tamponade and electromechanical dissociation, moderate-to-severe pericardial effusion increases the mortality risk. Those patients without initial cardiac tamponade, while at a lower rate of mortality, should still be followed, as late rupture may still occur. [13] The absence of pericardial effusion on echocardiography has high negative predictive value. If the ability to obtain transthoracic echocardiograms is limited in patients receiving mechanical ventilation, transesophageal echocardiography can assist in confirming VFWR.MRI provides superior image quality and permits identification of the site and anatomy of a ventricular pseudoaneurysm (ie, ruptured LV restrained by the pericardium with enclosed clot). However, MRI is of limited use in the acute setting because of the time involved and nonportability of imaging units.Treatment of VFWRThe most important prevention strategy is early reperfusion therapy, with percutaneous coronary intervention (PCI) being the preferred modality. Fibrinolytic therapy is associated with overall decreased risk of VFWR; however, its use more than 14 hours after STEMI onset can increase the risk of early rupture. [14, 15]The standard treatment for VFWR is emergency surgical repair after hemodynamic stability is achieved. Patients may first need intravenous fluids, inotropic agents, and emergency pericardiocentesis.Pifarré and associates recommended the deployment of an intra-aortic balloon pump to decrease systolic afterload and improve diastolic myocardial perfusion. [16]Several surgical techniques have been applied, including infarctectomy, adhering with biologic glue patches made of polyethylene terephthalate polyester fiber (Dacron; DuPont, Wilmington, DE) or polytetrafluoroethylene fluoropolymer resin (Teflon; DuPont); and use of pledgeted sutures without infarctectomy.The mortality rate is significantly high and largely depends on the patient's preoperative hemodynamic status. Early diagnosis, rapid institution of the measures described above to achieve hemodynamic stability, and prompt surgical repair can improve survival rates. A follow-up to the Acorn randomized trial demonstrated long-term improvement in left ventricular structure and function after mitral valve surgery for as long as 5 years. These data provide evidence supporting mitral valve repair in combination with the Acorn CorCap device for patients with nonischemic heart failure with severe left ventricular dysfunction who have been medically optimized yet remain symptomatic with significant mitral regurgitation. [17]Overview of ventricular septal ruptureVSR is an infrequent but life-threatening complication of AMI. Despite optimal medical and surgical treatment, patients with VSR have a high in-hospital mortality rate. During the prethrombolytic era, VSRs occurred in 1-3% of individuals with MIs. The incidence declined with thrombolytic therapy (to 0.2-0.34%) because of improvements in reperfusion and myocardial salvage. The bimodal distribution of VSR is characterized by a high incidence in the first 24 hours, with another peak on days 3-5 and rarely more than 2 weeks after AMI.In patients receiving thrombolytics, the median time from the onset of symptoms of AMI to septal rupture was 1 day in the Global Utilization of Streptokinase and TPA [tissue plasminogen activator] for Occluded Coronary Arteries (GUSTO-I) trial [18] and 16 hours in the Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock? (SHOCK) trial. [19]Risk factors for septal rupture include advanced age (>65 y), female sex, single-vessel disease, extensive MI, and poor septal collateral circulation. [20, 21] Before the advent of thrombolytics, hypertension and absence of a history of angina were risk factors for VSR. Extensive infarct size and RV involvement are other known risk factors for septal rupture.In patients with AMI without reperfusion, coagulation necrosis develops within 3-5 days after infarction. Neutrophils migrate to the necrotic zone and undergo apoptosis, release lytic enzymes, and hasten the disintegration of necrotic myocardium. Some patients have infarcts with large intramural hematomas, which dissect into the tissue and result in early septal rupture. The size of the septal rupture ranges from a few millimeters to several centimeters.VSR is categorized as simple or complex depending on its length, course, and location. In simple septal rupture, the perforation is at the same level on both sides of the septum, and a direct through-and-through communication is present across the septum. A complex septal rupture is characterized by extensive hemorrhage with irregular, serpiginous tracts in the necrotic tissue.Septal ruptures are most common in patients with large anterior MIs due to occlusion of the LAD artery causing extensive septal infarcts. These infarcts are associated with ST-segment elevations and Q waves in inferior leads (II, III, aVF) and these ECG changes are therefore more commonly seen in septal ruptures. [22] These ruptures are generally apical and simple.Septal ruptures in patients with inferior MI occur relatively infrequently. These ruptures involve the basal inferoposterior septum and are often complex.Clinical presentation of VSRSymptoms of VSR complicating AMI include chest pain, shortness of breath, hypotension, biventricular failure, and shock within hours to days. Patients often present with a new, loud, and harsh holosystolic murmur. This murmur is loudest along the lower left sternal border and is associated with a palpable parasternal systolic thrill. RV and LV S3 gallops are common.In patients with cardiogenic shock complicating septal rupture, the murmur and thrill may be difficult to identify. In contrast, patients with acute MR often have a soft systolic murmur at the apex without a thrill.Diagnosis of VSREchocardiography with color flow Doppler imaging is the diagnostic tool of choice for identifying a VSR. (See the image below.) Its sensitivity and specificity have been reported to be as high as 100%. In addition, it can be used for the following:Define the site and size of septal ruptureAssess the LV and RV functionEstimate the RV systolic pressureQuantify the left-to-right shuntCardiac catheterization is usually required to confirm the diagnosis, quantitate the degree of left-to-right shunt, differentiate VSR from other conditions (eg, mitral regurgitation), plus visualize the coronary arteries.Modified 2-dimensional (top) echocardiogram and coModified 2-dimensional (top) echocardiogram and color flow Doppler image (bottom). Apical 4-chamber views show a breach in the interventricular septum and free communication between ventricles through a large apical septum ventricular septal defect in a patient who recently had an anterior myocardial infarction.View Media GalleryIn patients with VSR, right-heart catheterization shows a step-up in oxygen saturation from the right atrium to the RV; in contrast, no step-up in oxygen saturation occurs among patients with MR. The presence of large V waves in the pulmonary capillary wedge tracing supports the diagnosis of severe acute MR.Left ventriculography can also be used to identify the site of ventricular rupture (see Cardiac Catheterization [Left Heart]). However, this study is usually unnecessary after a good-quality echocardiographic and Doppler examination is conducted.Treatment of VSRThe key to management of VSR is prompt diagnosis and an aggressive approach to hemodynamic stabilization, angiography, and surgery. The optimal approach includes hemodynamic stabilization with the administration of oxygen and mechanical support with use of an intra-aortic balloon pump, as well as the administration of vasodilators (to reduce afterload and thus LV pressure and the left-to-right shunt), diuretics, and inotropic agents.Cardiac catheterization is needed to define the coronary anatomy; this is followed by urgent surgical repair.Medical therapy is intended only for temporary stabilization before surgery, as most patients' conditions deteriorate rapidly and they die in the absence of surgical intervention. In the GUSTO-I trial, the 30-day mortality rate was lower in patients with VSR who underwent surgical repair than in patients treated medically (47% vs 94%), as was the 1-year mortality rate (53% vs 97%). [18] Lemery et al reported a 30-day survival rate of 24% in patients treated medically compared with 47% in those treated surgically. [23]Current guidelines of the American College of Cardiology/American Heart Association for the treatment of patients with septal rupture complicating AMI highlight urgent surgical intervention, regardless of their clinical status. [24] Surgical management of septal rupture includes the following elements:Prompt establishment of hypothermic cardiopulmonary bypassAn approach to the septal rupture through the infarct area and the excision of all necrotic, friable margins of the septum and ventricular walls to avoid postoperative hemorrhage, residual septal defect, or bothReconstruction of the septum and ventricular walls by using prosthetic material and preservation of the geometric configuration of the ventricles and heart functionPercutaneous closure of septal rupture is a relatively new approach, one used in select patients as an alternative to surgical repair or for the acute stabilization of critically ill patients. However, percutaneous closure is currently unavailable in many institutions, and no long-term outcome data are available.Several studies failed to show a relationship between perioperative mortality and concomitant coronary revascularization (coronary artery bypass grafting). Patients with cardiogenic shock due to septal rupture have the poorest outcome. In the SHOCK trial, the in-hospital mortality rate was higher in patients with cardiogenic shock due to septal rupture (87.3%) than in patients with cardiogenic shock from all other causes (59.2% with pure LV failure and 55.1% with acute MR). [19, 25]In patients who survive surgical repair, the rate of recurrent or residual septal defect is reported to be about 28%, and the associated mortality rate is high.Repeat surgical intervention is indicated in patients who have clinical heart failure or a pulmonary-systemic fraction greater than 2.Overview of acute mitral regurgitationMR is a common complication of AMI that results from local and global LV remodeling and that is an independent predictor of heart failure and death. MR typically occurs 7-10 days after an AMI, though this onset may vary according to the mechanism of MR. Papillary muscle rupture resulting in MR occurs within 1-14 days (median, 1 d).Mild-to-moderate MR is often clinically silent and detected on Doppler echocardiography performed during the early phase of AMI. In such cases, MR rarely causes hemodynamic compromise.Speckle tracking and 3-dimensional echocardiography proved to be important imaging tools in assessing reverse LV remodeling after degenerative mitral valve regurgitation surgery. Subtle regional preoperative changes in diastolic function of the septal and lateral wall could be preoperatively identified, aiding in optimizing the referral timing and recognizing potential culprits as indicators of disease recurrence after mitral repair. [26]Severe acute MR that results from the rupture of papillary muscles or chordae tendineae results in abrupt hemodynamic deterioration with cardiogenic shock. Rapid diagnosis, hemodynamic stabilization, and prompt surgical intervention are needed because acute severe MR is associated with a high mortality rate.The reported incidence of MR may vary because of several factors, including the diagnostic methods used, the presence or absence of heart failure, the degree of MR reported, the type of therapy rendered, and the time from infarct onset to testing.During the GUSTO-I trial, the incidence of MR in patients receiving thrombolytic therapy was 1.73%. [18] The SHOCK trial, which included MI patients presenting with cardiogenic shock, noted a 39.1% incidence of moderate to severe MR. [27] Kinn et al reported that reperfusion with angioplasty resulted in an 82% decrease in the rate of acute MR, as compared with thrombolytic therapy (0.31% vs 1.73%). [28]Risk factors for MR are advanced age, female sex, large infarct, previous AMI, recurrent ischemia, multivessel coronary artery disease, and heart failure.Several mechanisms can cause MR after AMI. Rupture of the papillary muscle is the most commonly reported mechanism.Such rupture occurs in 1% of patients with AMI and frequently involves the posteromedial papillary muscle rather than the anterolateral papillary muscle, as the former has a single blood supply versus the dual supply for the latter. Papillary muscle rupture may lead to flailing or prolapse of the leaflets, resulting in severe MR. Papillary muscle dysfunction due to scarring or recurrent ischemia may also lead to MR in the subacute and chronic phases after MI; this condition can resolve spontaneously.Large posterior infarctions produce acute MR due to asymmetric annular dilation and altered function and geometry of the papillary muscle.Clinical presentation of MRPatients with functional mild or moderate MR are often asymptomatic. The severity of symptoms varies depending on ventricular function. Clinical features of acute severe MR include shortness of breath, fatigue, a new apical holosystolic murmur, flash pulmonary edema, and shock.The new systolic murmur may be only early-to-mid systolic, not holosystolic. It may be soft or even absent because of the abrupt rise in left atrial pressure, which lessens the pressure gradient between the left atrium and the LV, as compared with chronic MR. The murmur is best heard at the apex rather than the lower left sternal border, and it is uncommonly associated with a thrill. S3 and S4 gallops are expected.Diagnosis of MRThe clinician cannot rely on a new holosystolic murmur to diagnose MR or assess its severity because of the variable hemodynamic status. In a patient with AMI who presents with a new apical systolic murmur, acute pulmonary edema, and cardiogenic shock, a high index of clinical suspicion for severe MR is the key to diagnosis.Chest radiography may show evidence of pulmonary edema in the acute setting without clinically significant cardiac enlargement.Echocardiography with color flow Doppler imaging is the standard diagnostic tool for detecting MR. Transthoracic echocardiography is the preferred initial screening tool, but transesophageal echocardiography is invaluable in defining the severity and exact mechanism of acute MR, especially when suspicion for papillary muscle rupture is high. Cardiac catheterization should be performed in all patients to determine the extent and severity of coronary artery disease.Treatment of MRDetermination of hemodynamic stability, elucidation of the exact mechanism of acute MR, and expedient therapy are all necessary for a favorable outcome. Medical management includes afterload reduction with the use of diuretics, sodium nitroprusside, and nitrates in patients who are not hypotensive.In patients who have hemodynamic compromise, intra-aortic balloon counterpulsation should be deployed rapidly. This intervention usually substantially reduces afterload and regurgitant volume, improving cardiac output in preparation for surgical repair. Without surgical repair, medical therapy alone in patients with papillary muscle rupture results in inadequate hemodynamic improvement and a poor short-term prognosis.Emergency surgical intervention is the treatment of choice for papillary muscle rupture. Surgical approaches may include mitral valve repair or replacement. In the absence of papillary muscle necrosis, mitral valve repair improves the survival rate more than mitral valve replacement does. This difference is because the subvalvular apparatus is usually preserved. Mitral valve repair also eliminates complications related to malfunction of the prosthesis.In patients with extensive necrosis of papillary muscle and/or ventricular free wall, mitral valve replacement is the preferred modality. Coronary artery bypass grafting (CABG) performed at the time of surgery was shown in one study to improve short- and long-term survival. [29]The only situation in which emergency surgery can safely be avoided is in the case of intermittent MR due to recurrent ischemia. In these patients, successful myocardial revascularization may be effective. This procedure is accomplished by means of either angioplasty or coronary artery bypass grafting.Left Ventricular AneurysmOverview of LVALeft ventricular aneurysm (LVA) is defined as a localized area of myocardium with abnormal outward bulging and deformation during both systole and diastole. The rate of LVAs after AMI is approximately 3-15%. Risk factors for LVA after AMI include female sex, total occlusion of the LAD artery, single-vessel disease, and absence of previous angina.More than 80% of LVAs affect the anterolateral wall; these are usually associated with total occlusion of the LAD. The posterior and inferior walls are less commonly affected. LVAs generally range from 1-8 cm. Histologically, LVAs are composed of fibrous scar that is notably thinned. This scar is clearly delineated from the adjacent ventricular muscle on microscopic examination.A history of MI and third or fourth heart sounds are common findings from the patient's history and physical examination.Diagnosis of LVAThe chest radiograph may reveal an enlarged cardiac silhouette.Electrocardiography is characterized by ST elevation that persists several weeks after AMI and that appears in the same leads as those showing the acute infarct. Echocardiography is 93% sensitive and 94% specific for detection of LVA (see the image below), but cardiac catheterization remains the standard for establishing the diagnosis.Parasternal long-axis view of the left ventricle dParasternal long-axis view of the left ventricle demonstrates a large inferobasal aneurysm. Note the wide neck and base of the aneurysm.View Media GalleryTreatment of LVAPatients with small or clinically insignificant aneurysms can be treated conservatively with close follow-up. Medical therapy generally consists of the use of angiotensin-converting enzyme (ACE) inhibitors, which reduce afterload, infarct extension, and LV remodeling. Anticoagulation is required when patients have severe LV dysfunction and/or thrombus in the LV or aneurysm.Surgical resection of the LVA is indicated if severe heart failure, ventricular tachyarrhythmias refractory to medical treatment, or recurrent thromboembolism is present.Miscellaneous ComplicationsLeft ventricular mural thrombusLVMT is a well-known complication of AMI and frequently develops after anterior infarcts of the LV wall. The incidence of LVMT as a complication of AMI ranges from 20-40% and may reach 60% in patients with large anterior-wall AMIs who are not treated with anticoagulant therapy. LVMT is associated with a high risk of systemic embolization. Anticoagulant therapy may substantially decrease the rate of embolic events by 33% compared with no anticoagulation.Factors contributing to LVMT formation include LV regional-wall akinesia or dyskinesia with blood stasis, injury to and inflammation of the endocardial tissue that provides a thrombogenic surface, and a hypercoagulable state. The most common clinical presentation of patients with LVMT complicating an MI is stroke. Most episodes occur within the first 10 days after AMI. Physical findings depend on the site of embolism.Transthoracic echocardiography remains the imaging modality of choice and is 92% sensitive and 88% specific for detecting LVMT (see the image below). Management of LVMT includes heparin treatment followed by oral warfarin therapy for 3-6 months. In patients with LVAs, lifelong anticoagulation may be appropriate if a mural clot persists.Apical 2-chamber view depicts a large left ventricApical 2-chamber view depicts a large left ventricular apical thrombus with mobile extensions.View Media GalleryPericarditisThe incidence of early pericarditis after MI is approximately 10%, and this complication usually develops within 24-96. Pericarditis is caused by inflammation of pericardial tissue overlying infarcted myocardium. The clinical presentation may include severe chest pain, usually pleuritic, and pericardial friction rub.The key ECG change is diffuse ST-segment elevation in all or nearly all of leads. Echocardiography may reveal a small pericardial effusion. The mainstay of therapy usually includes aspirin and nonsteroidal anti-inflammatory drugs (NSAIDs). Colchicine may be beneficial in patients with recurrent pericarditis.Post-MI syndrome (Dressler syndrome)Before the era of reperfusion, the incidence of post-MI syndrome ranged from 1-5% after AMI, but this rate has dramatically declined with the advent of thrombolysis and coronary angioplasty.Although the exact mechanism has yet to be elucidated, post-MI syndrome is considered to be an autoimmune process. Clinical features include fever, chest pain, and other signs and symptoms of pericarditis occurring 2-3 weeks after AMI. Management involves hospitalization and observation for any evidence of cardiac tamponade. Treatment comprises rest, use of NSAIDs, and/or steroids in patients with recurrent post-MI syndrome with disabling symptoms."Off-hour" MI admissionsDespite the perception that patients with an acute MI admitted during off hours have higher rates of death compared to those admitted during regular hours, a multivariate analyses of outcomes data at the Mayo Clinic for weekends, nights, and holidays admissions of patients with acute MI who underwent percutaneous coronary interventions found no significant association for inpatient mortality, 30-day mortality, or 30-day readmissions, nor were there any differences in findings between those with or without ST-elevation MI (STEMI, non-STEMI). [30] However, the investigators did find a significant association between off-hour admissions of these patients with composite major complications as well as emergent coronary artery bypass graft surgery, ventricular arrhythmia, cerebrovascular events, and hemorrhage (gastrointestinal, retroperitoneal, intracranial). |
|
|
|
Inferior mi manifestations |
Q |
|
|
|
Q Does Dipyridamole therapy reduces re infarction within the first year |
There was no evidence that dipyridamole alone was more efficacious than aspirin. Further trials comparing the effects of the combination of dipyridamole with aspirin with aspirin alone are justified. |
CONCLUSIONS: This review found that, for patients who presented with arterial vascular disease, there was no evidence that dipyridamole, in the presence or absence of an other antiplatelet drug (chiefly aspirin) reduced the risk of vascular death, though it may reduce the risk of further vascular events. However, this benefit was found in only a single large trial and only in patients presenting after cerebral ischaemia. |
|
|
Q 16. Characteristc features of pulmonary embolism includeReduced plasma lactate levels An increase in serum troponin levelsAn arterial pH less than 7.2Blood gases show increased pCO2 on airNormal D dimer levels
|
Q |
|
|
|
What is S1Q3T3 |
Q |
|
|
|
Characteristc features of pulmonary embolism include Reduced plasma lactate levels An increase in serum troponin levels An arterial pH less than 7.2 💡 Blood gases show increased pCO2 on air Normal D dimer levels
|
Patients with pulmonary embolism and elevated plasma lactate level are at high risk of death and adverse outcome, independent of shock or hypotension, or right-sided ventricular dysfunction or injury markers. |
The diagnosis of pulmonary embolism (PE) requires objective testing. However, all imaging techniques have their own limitations and costs and cannot be performed in every patient with suspected PE. After decades of unfruitful research, several laboratory tests have been evaluated for suspected PE, the most promising being the D-dimer test. As a general rule, the specificity of D-dimers is too low to confirm PE. Conversely, several (but not all) D-dimer assays have a high sensitivity for diagnosing PE. Outcome studies indicate that the Vidas D-dimer and SimpliRED D-dimer can be used safely to withdraw anticoagulation when the pretest probability of PE is low (SimpliRED) or when it is low or moderate (Vidas). These results may however not apply to other D-dimer assays and clinicians should know the characteristics of the test used in their hospital. Blood gas analysis does not have sufficient sensitivity and specificity to confirm or exclude PE, but it may be used to evaluate the clinical probability of PE before other testing is done. The diagnostic value of the alveolar dead space fraction in patients with suspected PE is currently investigated. Initial data suggest that it needs to be combined with a D-dimer test to safely exclude PE. Brain natriuretic peptide and cardiac troponin have limited usefulness for diagnosing PE, but both tests may identify patients with a poor prognosis, in whom more aggressive treatment may be warranted. |
|
|
RISK ASSESSMENT FOR ANTICOAGULATION FOR AF criteria |
Q Moderate High Low |
|
|
|
19. In a pt with type 2 DM WOF co existing clinical situations are a CI to the use of a tthazolidinedionePresence of bladder cancerProliferative diabetic retinopathySvere CCFPVDPresence of bowel cancer
|
• Bladder cancer o Pioglitazone may be linked to an increased risk of bladder cancer o Do not prescribe for patients with active bladder cancer Do not initiate pioglitazone in patients with NYHA Class III or IV heart failure (see Black Box Warnings) Hypersensitivity to alogliptin or pioglitazone, including anaphylaxis, angioedema, or severe cutaneous adverse reactions including Stevens-Johnson syndrome |
|
|
|
Diclofenac sodium in treatment of gout |
Sudden (acute) attacks of gout are usually treated with non- steroidal anti-inflammatory drugs (NSAIDs). NSAIDs such as diclofenac, naproxen or ibuprofen relieve pain and help to reduce the redness and swelling. ... NSAIDs should be used at the lowest possible dose for as short a duration as possible. |
|
|
|
Management of stable angina - drugs |
Summary Stable angina pectoris is characterised by typical exertional chest pain that is relieved by rest or nitrates.Risk stratification of patients is important to define prognosis, to guide medical management and to select patients suitable for revascularisation. Medical treatment aims to relieve angina and prevent cardiovascular events. Beta blockers and calcium channel antagonists are first-line options for treatment. Short-acting nitrates can be used for symptom relief. Low-dose aspirin and statins are prescribed to prevent cardiovascular events. Angina is caused by myocardial ischaemia. Chronic stable angina has a consistent duration and severity, and is provoked by a predictable level of exertion. It can also be provoked by emotional stress. The pain is relieved by rest or short-acting nitrates. The aim of medical therapy is to minimise symptoms and retard disease progression. This requires lifestyle modification as well as drug treatment. The diagnosis of angina is usually suspected from a thorough history and examination. Patients should have an ECG and undergo assessment for cardiovascular risk factors such as diabetes and hyperlipidaemia. An echocardiograph can help with the assessment of left ventricular function. Once the clinical diagnosis of stable coronary artery disease is established, the patient’s risk of future cardiovascular events is evaluated. Risk stratification In patients with stable coronary artery disease the risk of cardiovascular mortality may be predicted by clinical and demographic variables. These include gender,9 left ventricular function,8,9 the provocation of myocardial ischaemia with stress testing,10,11 and the severity of coronary artery disease seen on angiography Patients at high risk of cardiovascular events may need revascularisation as well as medical therapy. Clinical evaluationThe history, examination, ECG and laboratory tests provide important prognostic information. Increasing age, chronic kidney disease, diabetes, hypertension, current smoking, previous myocardial infarction, hypercholesterolaemia and heart failure are predictive of adverse outcomes Echocardiography provides information about left ventricular function, and regional wall motion abnormalities that may be related to infarction or ischaemia. In patients with stable coronary artery disease, left ventricular ejection fraction is the strongest predictor of long-term survival. Stress testing on a treadmill or bicycle is recommended for patients with normal resting ECGs who can exercise. Symptoms such as chest discomfort and dyspnoea, exercise workload, blood pressure response and ECG changes consistent with ischaemia are recorded as the patient exercises. Abnormalities present at rest such as atrial fibrillation, left ventricular hypertrophy, intraventricular conduction abnormalities and ECG changes related to electrolyte imbalance or digoxin will result in more frequent false-positive results. Stress testing is also used to evaluate the efficacy of revascularisation and medical treatment, and to direct the prescription of exercise. Exercise or pharmacological stress echocardiography may be necessary to demonstrate ischaemic changes in left ventricular systolic function in patients whose resting ECGs abnormal or unable to be interpreted (because of left bundle branch block, paced rhythm). Exercise echocardiography provides information about cardiac structure and function, exercise workload, heart rate and rhythm and blood pressure response. Pharmacological testing may be necessary in patients who cannot exercise. Myocardial perfusion scintigraphy is an alternative for those with uninterpretable ECGs or inability to exercise. Imaging of coronary arteriesComputed tomography (CT) of the coronary arteries without contrast injection can show coronary calcification,17 although correlation with the degree of luminal narrowing is poor.Intravenous injection of a contrast agent allows visualisation of the vessel lumen. The severity and extent of the lesions determine the risk of a cardiovascular event (Table 1).12,16,18-20 CT angiography exposes patients to radiation. It should be reserved for those who are not overweight, without excessive coronary calcium (Agatston score <400) and who are in sinus rhythm with resting heart rates of 65 beats/minute or less, with or without medication.Table 1Table 1Risk stratification by CT coronary angiography 12,16,18-20If patients have a high risk of cardiovascular events or if their symptoms are not adequately controlled, invasive coronary angiography may be indicated. It helps define prognosis5 and options for revascularisation. The 12-year survival rate in medically treated patients is 74% for single-vessel disease, 59% for two-vessel disease and 50% for three-vessel coronary disease.12 Severe stenosis of the left main coronary artery or proximal left anterior descending artery has a poor prognosis if not revascularised.8 Conversely, the exclusion of significant obstructive disease on angiography is reassuring.19Lifestyle modificationThe management of cardiovascular risk factors plays an important role in the overall care of patients with chronic stable angina (Fig.). Modifiable cardiovascular risk factors include hypertension, hypercholesterolaemia, smoking, diabetes, obesity and sedentary lifestyle. Regular exercise, a healthy diet and maintenance of ideal weight reduce the risk of adverse cardiovascular events. Smoking is a strong and independent risk factor for coronary artery disease so efforts to quit should be encouraged and supported. Control of blood pressure and diabetes is paramount to reducing cardiovascular morbidity and mortality. Patients should be screened for sleep apnoea. Annual influenza vaccination is recommended.21,22FigFigManagement of chronic stable anginaPrevention of cardiovascular eventsLow-dose aspirin reduces major cardiac events by up to 30% and should be prescribed to patients with coronary artery disease.3 Clopidogrel is an alternative option for patients intolerant of aspirin. Patients with established coronary artery disease should be prescribed statin therapy irrespective of their lipid profile to slow the progression or even promote regression of coronary atherosclerosis.4 Angiotensin converting enzyme (ACE) inhibitors should be prescribed for patients with stable angina, particularly those who have hypertension, left ventricular dysfunction, diabetes6 or chronic kidney disease. Adverse effects include a persistent cough, hyperkalaemia and, rarely, angioedema. Angiotensin receptor antagonists may be used for those who do not tolerate ACE inhibitors.3 Drug therapyThe aim of drug therapy (Table 2)2,3,5,23 is to minimise symptoms and prevent progression of coronary artery disease. Short-acting nitrates are prescribed to relieve acute symptoms or anticipated angina. Drug therapy aims to reduce myocardial oxygen demand or increase coronary blood supply. The choice of drugs is influenced by factors such as comorbidities, tolerance and adverse effects.Table 2Table 2 Drugs for angina Beta blockers Beta blockers are first-line therapy to reduce angina and improve exercise tolerance by limiting the heart rate response to exercise.3,5 Although they reduce the risk of cardiovascular death and myocardial infarction by 30% in post-infarct patients, their benefits in those with stable coronary artery disease are less certain.3,24 The drugs most widely used for angina in the context of normal left ventricular function are the beta1-selective drugs such as metoprolol and atenolol.Adverse effects include fatigue, altered glucose, bronchospasm, bradycardia, impotence and postural hypotension. Switching to a less lipophilic beta blocker such as atenolol may alleviate symptoms such as insomnia or nightmares. They are usually well tolerated in patients with emphysema who have predominantly fixed airways disease. Beta blockers should not be stopped abruptly due to the risk of rebound hypertension or ischaemia. Calcium channel antagonistsCalcium channel antagonists improve symptoms of angina via coronary and peripheral vasodilation. They are indicated for those who cannot tolerate or have insufficient control of ischaemic symptoms on beta blockers alone.Non-dihydropyridine drugs such as verapamil and diltiazem also reduce heart rate and contractility. Verapamil has comparable antianginal activity to metoprolol and can be useful for treatment of supraventricular arrhythmias and hypertension. However, verapamil should be avoided in patients taking beta blockers owing to the risk of heart block, and in those with heart failure because of its negative inotropic effect. Diltiazem has a low adverse effect profile with a modest negative inotropic effect. Care should be taken when prescribing in combination with a beta blocker and in patients with left ventricular dysfunction.The dihydropyridines such as amlodipine, felodipine and lercanidipine have greater vascular selectivity and minimal negative inotropic properties. They are therefore safer in patients with left ventricular dysfunction. Amlodipine is an effective once-daily antianginal drug that can be used in combination with a beta blocker. Long-acting nifedipine is a proven antianginal drug and is most effective when used in conjunction with a beta blocker.25Contraindications to nifedipine use include severe aortic stenosis, obstructive cardiomyopathy and heart failure. Short-acting nifedipine is rarely used as monotherapy due to reflex tachycardia, which can worsen ischaemia and has been associated with a dose-related increase in mortality. It should therefore be avoided.NitratesSublingual glyceryl trinitrate tablets or nitroglycerin spray remain the treatment of choice for rapid relief of acute symptoms and anticipated angina. Sublingual glyceryl trinitrate tablets are absorbed in the sublingual mucosa and take effect within a couple of minutes. The tablet can be discarded with resolution of chest pain to minimise adverse effects such as headache. Glyceryl trinitrate spray is equally effective and, due to its longer shelf-life, is more convenient for those with infrequent symptoms of angina.Isosorbide dinitrate undergoes hepatic conversion to mononitrate, resulting in an onset of action of 3–4 minutes. It can provide an antianginal effect for up to one hour. Less commonly it is used as a chronic antianginal drug but requires multiple dosing, and tolerance limits its usefulness. It is often used up to three times per day with a nitrate-free period of up to 14 hours to minimise tolerance.Long-acting nitrates such as oral isosorbide mononitrate or transdermal patches are effective in relieving angina and can improve exercise tolerance. Chronic nitrate therapy is limited by the development of nitrate tolerance. A nitrate-free period of at least eight hours may reduce this problem. The mechanism of nitrate tolerance is not well established but involves attenuation of the vascular effect of the drug rather than altered pharmacokinetics.26 A nitrate-free period restores the vascular reactivity of the vessel. Transdermal patches are generally used for 12 consecutive hours with a 12-hour nitrate-free period. There is no evidence that nitrates improve survival.Common adverse effects include headache, hypotension and light-headedness. Nitrates should not be prescribed for patients taking phosphodiesterase-5 inhibitors such as sildenafil due to the risk of profound hypotension. Other contraindications include severe aortic stenosis and hypertrophic cardiomyopathy.NicorandilNicorandil is a potassium channel activator that improves coronary flow as a result of both arterial and venous dilation. It may be used in addition to beta blockers and calcium channel antagonists to control angina or in patients who are intolerant of nitrates. Nicorandil has been shown to reduce cardiovascular events by 14% in patients with chronic stable angina.27 Its use has been associated with headaches, hypotension, painful ulcers and genital and gastrointestinal fistulae.28IvabradineIvabradine can be considered for patients intolerant of, or insufficiently responsive to, other drugs. It acts on If channels in the sinus node to lower the heart rate of patients in sinus rhythm without affecting blood pressure, conduction or myocardial contractility.29 Ivabradine has been shown to reduce a composite primary end point of cardiovascular death and hospitalisation with myocardial infarction or heart failure. However, a recent placebo-controlled trial involving 19 102 patients with stable coronary artery disease found that adding ivabradine to standard therapy did not improve a composite outcome of death from cardiovascular causes, or non-fatal myocardial infarction.30 Ivabradine has been used in combination with beta blockers.31PerhexilinePerhexiline promotes anaerobic metabolism of glucose in active myocytes. Its use is limited by a narrow therapeutic window and high pharmacokinetic variability.23 Given its potential for toxic effects such as peripheral neuropathy and hepatic damage, it is usually reserved for patients whose angina is refractory to other therapies. It may be used safely with conscientious monitoring of clinical effects and regular measurement of plasma drug concentrations.32ConclusionStable angina is typically provoked by exertion and relieved by rest or nitrate therapy.2 Risk stratification should be done to define prognosis, guide management and select appropriate patients for revascularisation.3,5,19 The aims of medical therapy are to control symptoms, improve quality of life and prevent cardiovascular events.2,5 Beta blockers and calcium channel antagonists remain first-line options for treatment. Short-acting nitrates can be used for symptoms.FootnotesConflict of interest: none declaredArticle informationAust Prescr. 2015 Aug; 38(4): 131–136.Published online 2015 Aug 3. doi: 10.18773/austprescr.2015.042PMCID: PMC4653970PMID: 26648642Yong Wee, Advanced trainee in cardiology, Kylie Burns, Cardiology fellow, and Nicholas Bett, CardiologistHeart Lung Institute, Prince Charles Hospital, BrisbaneCopyright noticeThis article has been cited by other articles in PMC.Articles from Australian Prescriber are provided here courtesy of NPS MedicineWiseReferences1. Australian Institute of Health and Welfare. Cardiovascular disease: Australian facts 2011. Cardiovascular disease series no 35. Cat. No. CVD 53. Canberra: AIHW; 2011. www.aihw.gov.au/publication-detail/?id=10737418510 [cited 2015 Jul 1]2. Abrams J. Clinical practice. Chronic stable angina. N Engl J Med 2005;352:2524-33. [PubMed]3. Montalescot G, Sechtem U, Achenbach S, Andreotti F, Arden C, Budaj A, et al. Task Force Members. ESC Committee for Practice Guidelines. Document Reviewers . 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J 2013;34:2949-3003. [PubMed]4. Catapano AL, Reiner Z, De Backer G, Graham I, Taskinen MR, Wiklund O, et al. European Society of Cardiology (ESC) European Atherosclerosis Society (EAS) . ESC/EAS Guidelines for the management of dyslipidaemias: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011;217:3-46. [PubMed]5. Fihn SD, Gardin JM, Abrams J, Berra K, Blankenship JC, Dallas AP, et al. American College of Cardiology Foundation. American Heart Association Task Force on Practice Guidelines. American College of Physicians. American Association for Thoracic Surgery. Preventive Cardiovascular Nurses Association. Society for Cardiovascular Angiography and Interventions. Society of Thoracic Surgeons . 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2012;60:e44-164. [PubMed]6. Heart Outcomes Prevention Evaluation Study Investigators . Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy [Erratum in: Lancet 2000;356:860]. Lancet 2000;355:253-9. [PubMed]7. Norhammar A, Lagerqvist B, Saleh N. Long-term mortality after PCI in patients with diabetes mellitus: results from the Swedish Coronary Angiography and Angioplasty Registry. EuroIntervention 2010;5:891-7. [PubMed]8. Emond M, Mock MB, Davis KB, Fisher LD, Holmes DR, Jr, Chaitman BR, et al. Long-term survival of medically treated patients in the Coronary Artery Surgery Study (CASS) Registry. Circulation 1994;90:2645-57. [PubMed]9. Hjemdahl P, Eriksson SV, Held C, Forslund L, Näsman P, Rehnqvist N. Favourable long term prognosis in stable angina pectoris: an extended follow up of the angina prognosis study in Stockholm (APSIS). Heart 2006;92:177-82. [PMC free article] [PubMed]10. Ashley EA, Myers J, Froelicher V. Exercise testing in clinical medicine. Lancet 2000;356:1592-7. [PubMed]11. Schinkel AF, Bax JJ, Geleijnse ML, Boersma E, Elhendy A, Roelandt JR, et al. Noninvasive evaluation of ischaemic heart disease: myocardial perfusion imaging or stress echocardiography? Eur Heart J 2003;24:789-800. [PubMed]12. Califf RM, Armstrong PW, Carver JR, D’Agostino RB, Strauss WE. 27th Bethesda Conference: matching the intensity of risk factor management with the hazard for coronary disease events. Task Force 5. Stratification of patients into high, medium and low risk subgroups for purposes of risk factor management. J Am Coll Cardiol 1996;27:1007-19. [PubMed]13. Prasad A, Rihal C, Holmes DR, Jr. The COURAGE trial in perspective. Catheter Cardiovasc Interv 2008;72:54-9. [PubMed]14. Rihal CS, Raco DL, Gersh BJ, Yusuf S. Indications for coronary artery bypass surgery and percutaneous coronary intervention in chronic stable angina: review of the evidence and methodological considerations. Circulation 2003;108:2439-45. [PubMed]15. Hemingway H, Crook AM, Feder G, Banerjee S, Dawson JR, Magee P, et al. Underuse of coronary revascularization procedures in patients considered appropriate candidates for revascularization. N Engl J Med 2001;344:645-54. [PubMed]16. Lin FY, Dunning AM, Narula J, Shaw LJ, Gransar H, Berman DS, et al. Impact of an automated multimodality point-of-order decision support tool on rates of appropriate testing and clinical decision making for individuals with suspected coronary artery disease: a prospective multicenter study. J Am Coll Cardiol 2013;62:308-16. [PubMed]17. Raggi P, Gongora MC, Gopal A, Callister TQ, Budoff M, Shaw LJ. Coronary artery calcium to predict all-cause mortality in elderly men and women. J Am Coll Cardiol 2008;52:17-23. [PubMed]18. Min JK, Dunning A, Lin FY, Achenbach S, Al-Mallah M, Budoff MJ, et al. CONFIRM Investigators . Age- and sex-related differences in all-cause mortality risk based on coronary computed tomography angiography findings results from the International Multicenter CONFIRM (Coronary CT Angiography Evaluation for Clinical Outcomes: An International Multicenter Registry) of 23,854 patients without known coronary artery disease. J Am Coll Cardiol 2011;58:849-60. [PubMed]19. Jacq L, Chabredier-Paquot C, Pezzano M, Caussin C, Habis M, Schaison F, et al. [Prognostic value of normal coronary angiography] [French.]. Ann Cardiol Angeiol (Paris) 2001;50:404-7. [PubMed]20. Ostrom MP, Gopal A, Ahmadi N, Nasir K, Yang E, Kakadiaris I, et al. Mortality incidence and the severity of coronary atherosclerosis assessed by computed tomography angiography. J Am Coll Cardiol 2008;52:1335-43. [PubMed]21. Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med 2003;348:1322-32. [PubMed]22. Ciszewski A, Bilinska ZT, Brydak LB, Kepka C, Kruk M, Romanowska M, et al. Influenza vaccination in secondary prevention from coronary ischaemic events in coronary artery disease: FLUCAD study. Eur Heart J 2008;29:1350-8. [PubMed]23. Horowitz JD, Mashford ML. Perhexiline maleate in the treatment of severe angina pectoris. Med J Aust 1979;1:485-8. [PubMed]24. Bangalore S, Bhatt DL, Steg PG, Weber MA, Boden WE, Hamm CW, et al. β-blockers and cardiovascular events in patients with and without myocardial infarction: post hoc analysis from the CHARISMA trial. Circ Cardiovasc Qual Outcomes 2014;7:872-81. [PubMed]25. Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation 1995;92:1326-31. [PubMed]26. Parker JD, Parker JO. Nitrate therapy for stable angina pectoris. N Engl J Med 1998;338:520-31. [PubMed]27. IONA Study Group . Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet 2002;359:1269-75. [PubMed]28. McDaid J, Reichl C, Hamzah I, Fitter S, Harbach L, Savage AP. Diverticular fistulation is associated with nicorandil usage. Ann R Coll Surg Engl 2010;92:463-5. [PMC free article] [PubMed]29. Fox K, Ford I, Steg PG, Tendera M, Ferrari R, BEAUTIFUL Investigators . Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet 2008;372:807-16. [PubMed]30. Fox K, Ford I, Steg PG, Tardif JC, Tendera M, Ferrari R, SIGNIFY Investigators . Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med 2014;371:1091-9. [PubMed]31. Tardif JC, Ponikowski P, Kahan T, ASSOCIATE Study Investigators . Efficacy of the I(f) current inhibitor ivabradine in patients with chronic stable angina receiving beta-blocker therapy: a 4-month, randomized, placebo-controlled trial. Eur Heart J 2009;30:540-8. [PMC free article] [PubMed]32. Ashrafian H, Horowitz JD, Frenneaux MP. Perhexiline. Cardiovasc Drug Rev 2007;25:76-97. [PubMed] |
|
|
|
For hypotensive patients with AF cardioversion is best elective or emergent |
Cardioversion Cardioversion may be performed electively or emergently to restore sinus rhythm in patients with new-onset AF. Cardioversion is most successful when initiated within 7 days after onset of AF. The need for cardioversion may be acute when AF is responsible for hypotension, heart failure, or angina.Pharmacologic agents or direct current energy can be used to cardiovert patients with AF. Pharmacologic cardioversion has the advantage of not requiring sedation or anesthesia, but the major disadvantage is the risk of ventricular tachycardia and other serious arrhythmias. |
|
|
|
Pneumonia Severity Is CRP associated with it |
CRP reference values The serum levels of CRP were strongly associated with bacterial infection and severe pneumonia. Therefore, the CRP level, along with other parameters, may be used as early indicators of severe pneumonia development. However, the efficiency of the CRP level alone to diagnose severe pneumonia was found to be limited. There was a trend for an association between the level of CRP on admission and the time to reach clinical stability (p <0.01). In conclusion, CRP may be valuable for revealing the development of complications in CAP. It may also be useful to assess the disease severity, thus being complementary to the assessment of the PSI. In our patients, high CRP levels were associated with a failure to reach clinical stability. |
|
|
|
Is empyema associated with community acquired pneumonia |
YEP |
|
|
|
Reference values |
Serum protein Serum albumin BUN Serum creatinine |
|
|
|
Q 29. Clostridium difficile infection It is the commonest diagnosed cause of diarrhoes in a hospitalized pt Severe cases can develop mega colon Microbiological diagnosis is made by stool toxin detection using PCR Macrolides are the antibiotics most commonly associated with causation of CDI For treatment IV vancomycin is equally effective as metronidazole |
Q |
|
|
|
Nice stroke |
NICE Technology Appraisal 90 41 Guidance This guidance applies to people who have had an occlusive vascular event, or who have symptomatic peripheral arterial disease. This guidance does not apply to people who have had, or are at risk of, a stroke associated with atrial fibrillation, or who require treatment to prevent occlusive events after coronary revascularisation or carotid artery procedures. 1.1 As part of the prevention of occlusive vascular events: 1.1.1 the combination of modified-release (MR) dipyridamole and aspirin is recommended for people who have had an ischaemic stroke or a transient ischaemic attack for a period of 2 years from the most recent event. Thereafter, or if MR dipyridamole is not tolerated, preventative therapy should revert to standard care (including long-term treatment with low-dose aspirin) 1.1.2 clopidogrel alone (within its licensed indications) is recommended for people who are intolerant of low-dose aspirin and either have experienced an occlusive vascular event or have symptomatic peripheral arterial disease. 1.2 For the purposes of this guidance, aspirin intolerance is defined as either of the following: • proven hypersensitivity to aspirin-containing medicines • history of severe dyspepsia induced by low-dose aspirin. 2 Clinical need and practice 2.1 Occlusive vascular events (OVEs) are the result of a reduction in blood flow related to the narrowing or blocking of an artery. Examples of such events include transient ischaemic attack (TIA), ischaemic stroke and myocardial infarction (MI).
NICE Technology Appraisal 90 52.2 Peripheral arterial disease (PAD) is also caused by narrowing of arteries. PAD may be asymptomatic but commonly presents with leg pain on walking (intermittent claudication). People with PAD are at high risk of OVEs, including MI, stroke or TIA. 2.3 Narrowing or blocking of an artery is usually caused by atherosclerosis and atherothrombosis. Atherosclerotic plaque formation within artery walls results from damage to the vascular endothelium caused by several insults working together over a long period. These include factors such as elevated low-density lipoproteins, cigarette smoking, hypertension and diabetes mellitus. Atherothrombosis is characterised by sudden atherosclerotic plaque disruption leading to platelet activation and thrombus (clot) formation. Such a thrombus may then block an artery, either locally or more distally by embolisation. 2.5 Ischaemic stroke and MI are associated with a high morbidity and mortality; 30% of people die from their first MI. After a stroke, approximately 23% of people die within 30 days, and of the initial stroke survivors, only 30–40% are alive after 3 years. Stroke is also the leading cause of disability in the UK and other Western countries, with about 25−30% of stroke survivors remaining permanently disabled. People who have had an OVE are at increased risk of recurrent events, and people with symptomatic atherosclerosis in one vascular bed are also at higher risk of subsequent events in other vascular beds. However, people who have had an MI are more likely to experience a recurrent MI than a stroke. People who have had a stroke are initially more likely to experience a recurrent stroke than to experience an MI. Within 1–5
6 years after a stroke, the risk of dying from a recurrent stroke becomes smaller than the risk of dying as a result of non-stroke cardiovascular events. 2.7 Antiplatelet agents have been shown to be effective in the prevention of recurrent OVEs. Aspirin is an antiplatelet agent and has been recommended by national clinical guidelines both for patients with coronary artery disease (including people who have survived an MI) and for people who have had an ischaemic stroke. For the prevention of cardiovascular events, daily doses of 75–150 mg aspirin have been recommended in national and international guidelines. For the prevention of ischaemic stroke, daily doses of 75-325 mg or 50–325 mg aspirin have been recommended (Royal College of Physicians guideline and European Stroke Initiative, respectively). The adverse effects of aspirin are related to bleeding complications and include haemorrhagic stroke and gastrointestinal haemorrhage. However, the benefits of antiplatelet treatment in people at moderate to high risk are considered to outweigh the risk of major bleeding. Clopidogrel is licensed for the prevention of atherothrombotic events in people who have had an MI (from a
NICE Technology Appraisal 90 7few days until less than 35 days), have had an ischaemic stroke (from 7 days until less than 6 months) or have established PAD. 3.1.2 Contraindications include severe liver impairment, active pathological bleeding and breastfeeding. Because of its antiplatelet activities, clopidogrel increases the risk of bleeding. For full details of side effects and contraindications, see the Summary of Product Characteristics. 3.1.3 The cost of treatment for 1 year at a dose of 75 mg daily is £460.29 (excluding VAT; British National Formulary, 46th edition). Costs may vary in different settings because of negotiated procurement discounts. 3.2 Dipyridamole 3.2.1 Dipyridamole (Boehringer Ingelheim) has both antiplatelet and vasodilating properties and is thought to inhibit the uptake of adenosine (a potent inhibitor of platelet activation and aggregation) into blood and vascular cells. Dipyridamole may also inhibit the breakdown of cyclic guanosine monophosphate. This guidance refers to the modified-release (MR) formulation of dipyridamole only, which is licensed for the secondary prevention of ischaemic stroke and TIAs, either alone (Persantin Retard, 200 mg dipyridamole twice daily) or in combination with aspirin (Asasantin Retard, 200 mg dipyridamole plus 25 mg aspirin twice daily). 3.2.2 Because of dipyridamole’s activity as a vasodilator, it should be used with caution in people with severe coronary artery disease, including unstable angina and/or recent MI, left ventricular outflow obstruction or haemodynamic instability (for example, decompensated heart failure). Contraindications and special warnings for the combination of MR dipyridamole with aspirin are associated with the product’s aspirin component and include active gastric or duodenal ulcers or bleeding disorders. 1 Clinical effectiveness 4.1.1 Clopidogrel 4.1.1.1 One RCT was identified that investigated the use of clopidogrel compared with aspirin in the prevention of OVEs. The CAPRIE (Clopidogrel versus Aspirin in Patients at Risk of Ischaemic Events) study included 19,185 patients with a diagnosis of ischaemic stroke, MI or symptomatic atherosclerotic PAD randomised to receive clopidogrel (75 mg/day) or aspirin (325 mg/day). The duration of follow-up ranged from 1 to 3 years (mean 1.9 years). The mean ages were similar in all groups, and all groups appeared to be well matched in terms of other prognostic indicators. The primary outcome was a composite endpoint of the first occurrence of ischaemic stroke, MI or vascular death. 4.1.1.2 The main analysis across the whole CAPRIE cohort found a lower incidence of the primary outcome with clopidogrel (9.8% over the whole study period) compared with aspirin (10.7%) (relative risk [RR] 0.92, 95% confidence interval [CI]: 0.85 to 1.00). There was statistically significant heterogeneity across the three qualifying subgroups (ischaemic stroke, MI and symptomatic PAD). When the results for each of the subgroups were analysed, there was a statistically significant effect only in patients with PAD (6.7% with clopidogrel, 8.6% with aspirin; RR 0.78, 95% CI: 0.66 to 0.92). The incidence of the primary outcome in people with ischaemic stroke was 13.4% with clopidogrel and 14.4% with aspirin (RR 0.93, 95% CI: 0.82 to 1.05) and for people with MI 9.3% with clopidogrel and 9% with aspirin
NICE Technology Appraisal 90 9aspirin (RR 1.03, 95% CI: 0.88 to 1.21). However, the data for these subgroups should be interpreted with caution because the trial was not powered to detect differences in treatment effects between the subgroups. 4.1.1.3 A further analysis determined the effect of treatment on the individual outcomes of the trial (preventing ischaemic stroke, MI or vascular death) across the whole CAPRIE cohort. Clopidogrel reduced the number of patients progressing to an MI (RR 0.81, 95% CI: 0.69 to 0.95). No statistically significant difference was found between clopidogrel and aspirin for ischaemic stroke (RR 0.95, 95% CI: 0.83 to 1.08) and vascular death (RR 0.92, 95% CI: 0.80 to 1.07) (incidence rates not available). 4.1.1.4 For the secondary outcomes (ischaemic stroke, MI, amputation or vascular death; vascular death; any stroke, MI or death from any cause; death from any cause), no statistically significant differences were found between clopidogrel and aspirin, despite point estimates favouring clopidogrel. 4.1.1.5 Overall, clopidogrel did not affect the number of bleeding disorders compared with aspirin. However, gastrointestinal haemorrhage rates were statistically significantly lower with clopidogrel than with aspirin. The incidences of rash and diarrhoea were higher in the clopidogrel group compared with the aspirin group, and the incidence of indigestion/nausea/vomiting was higher in the aspirin group.
NICE Technology Appraisal 90 104.1.2 MR dipyridamole 4.1.2.1 The European Stroke Prevention Study 2 (ESPS-2) was the only randomised controlled trial (RCT) identified that evaluated MR dipyridamole, aspirin, and MR dipyridamole combined with aspirin (aspirin/MR dipyridamole) compared with placebo. Patients included in the trial had experienced a TIA or an ischaemic stroke within the preceding 3 months. The study included 6602 patients randomised to aspirin (50 mg/day), aspirin/MR dipyridamole (50 mg/day plus 400 mg/day, respectively), MR dipyridamole (400 mg/day) or placebo. Patients were followed on treatment for 2 years. The mean ages of these four groups were similar and patients appeared to be well matched in terms of prognostic indicators. The primary outcomes were stroke, stroke and/or death, and death. 4.1.2.2 In the ESPS-2 study, aspirin (50 mg/day) alone was more effective than placebo, with an 18.1% reduction in the outcome of stroke compared with placebo. Compared with aspirin, MR dipyridamole treatment alone did not cause a reduction in the outcomes of stroke, death or any of the secondary outcomes. For the secondary outcome of MI, there was a small but statistically insignificant increase with MR dipyridamole compared with aspirin. 4.1.2.3 The ESPS-2 study found a lower incidence of stroke with aspirin/MR dipyridamole (9.5%) compared with aspirin alone (12.5%) (RR 0.76, 95% CI: 0.63 to 0.93). The effect on the outcome ‘stroke and/or death’ was considered uncertain, with incidence rates of 17.3% with aspirin/MR dipyridamole and 20% with aspirin alone (RR 0.87, 95% CI: 0.75 to 1.00). There was no difference between the two treatments for the outcome of death (RR 1.02, 95% CI: 0.84 to 1.23). Among the secondary outcomes, the following were statistically significantly reduced for the aspirin/MR dipyridamole combination compared with aspirin alone: stroke
NICE Technology Appraisal 90 11or TIA (18.1% with aspirin/MR dipyridamole, 22.6% with aspirin alone; RR 0.80, 95% CI: 0.70 to 0.92), other vascular event (1.3% with aspirin/MR dipyridamole, 2.3% with aspirin alone; RR 0.55, 95% CI: 0.33 to 0.94), fatal and non-fatal ischaemic events (12.5% with aspirin/MR dipyridamole, 16.1% with aspirin alone; RR 0.77, 95% CI: 0.65 to 0.92), and vascular events (14.9% with aspirin/MR dipyridamole, 19% with aspirin alone; RR 0.78, 95% CI: 0.67 to 0.91). Point estimates suggested a reduction in the secondary outcomes TIA, MI and vascular death, but these effects were not statistically significant. 4.1.2.4 The frequency of bleeding complications was significantly lower with MR dipyridamole (4.7%) than with aspirin (8.2%), and similar in the aspirin/MR dipyridamole (8.7%) and the aspirin (8.2%) groups. The frequencies of headache, diarrhoea, nausea and vomiting were significantly higher in both the aspirin/MR dipyridamole and MR dipyridamole groups compared with aspirin. 4.1.2.5 The ATTC reviewed 25 studies of aspirin in combination with dipyridamole. The authors of the ATTC meta-analysis and of a Cochrane Review concluded that the addition of dipyridamole to aspirin failed to clearly demonstrate additional reductions in serious vascular events. This assessment by the ATTC and the Cochrane Group relates mainly to studies conducted with the standard-release preparation of dipyridamole and not MR dipyridamole, which has a significantly different absorption profile compared with the standard-release preparation.4.2 Cost effectiveness 4.2.1 None of the previously published studies of cost effectiveness for MR dipyridamole or clopidogrel could be generalised to the UK setting and the NHS. The manufacturers (Boehringer Ingelheim and SSBMS, respectively) submitted new or updated models. The Assessment Group developed a new model based on the SSBMS model. NICE Technology Appraisal 90 124.2.2 The model submitted by Boehringer Ingelheim compared MR dipyridamole (with or without aspirin) and aspirin against placebo for the prevention of stroke. The model separated the stroke-survivor cohort into disabled (30.9%) and non-disabled (69.1%), and treatment was for each individual’s lifetime, including withdrawal rates seen in the relevant trial. The model considered only the first recurrent stroke. In this model, MR dipyridamole (alone) was less effective and more costly than aspirin. For the combination of aspirin and MR dipyridamole, the cost per quality-adjusted life year (QALY) gained compared with aspirin alone was £4207 for people with stroke and £9448 for people with TIA (5-year analysis). When the time horizon was extended to 30 years, the cost per additional QALY was £3655 for people with stroke and £2038 for people with TIA. 4.2.3 The model submitted by SSBMS compared clopidogrel with aspirin for the prevention of OVEs. Treatment was for 2 years, followed by lifetime treatment with aspirin. The time horizon of the model was 40 years. Adverse events associated with treatment were not modelled, and all stroke-qualifying patients began the model as non-disabled. This model resulted in a cost per QALY gained for clopidogrel of £14,525 compared with aspirin. In an additional analysis for each qualifying subgroup, the costs per additional QALY were £12,527 for the MI group, £15,896 for the stroke group and £17,218 for the PAD group. 4.2.4 The Assessment Group compared clopidogrel and MR dipyridamole (with or without aspirin) with aspirin in the prevention of OVEs. The analysis included both the cost effectiveness of lifetime treatment as well as 2-year treatment followed by lifetime treatment with aspirin (as in the SSBMS model). The cost effectiveness in each relevant qualifying subgroup (stroke, TIA, MI and PAD) was estimated separately, but the RRs for the outcomes were based on the overall results of the trials. The model used RRs for the progression to the individual outcomes (non-fatal stroke, non-fatal MI, vascular or non-vascular death) derived from ESPS-2 and CAPRIE. The time horizon of the analysis was 40 years. Because of the different licensed indications for clopidogrel andNICE Technology Appraisal 90 13MR dipyridamole (with or without aspirin), different combinations of treatments were compared in each subgroup by qualifying event. • Stroke: aspirin, clopidogrel, aspirin/MR dipyridamole and MR dipyridamole (alone). • TIA: aspirin, aspirin/MR dipyridamole and MR dipyridamole (alone). • MI or PAD: aspirin and clopidogrel. 4.2.5 In the Assessment Group analysis for people with stroke or TIA, lifetime treatment with aspirin/MR dipyridamole resulted in a cost per additional QALY of £26,432 and £12,458 for stroke or TIA, respectively, relative to treatment with aspirin. When treatment duration was reduced to 2 years (followed by treatment with aspirin alone for the remainder of the individual’s lifetime), these figures fell to £5500 and £2241. This analysis excluded the relative effects of antiplatelet therapies on non-vascular death. When the RR for non-vascular death was included in the modelling, aspirin/MR dipyridamole was dominated by aspirin alone in the lifetime treatment scenario, and for the 2-year treatment duration, the cost per additional QALY was £7968 for people with stroke and £4266 for people with TIA. Treatment with clopidogrel (stroke subgroup only) resulted in high cost per QALY figures compared with aspirin/MR dipyridamole (which was the next more cost-effective comparator) or was less effective and more costly than the other treatments (lifetime and 2-year treatment duration analysis). MR dipyridamole (alone) was less effective and more costly than aspirin (lifetime and 2-year treatment analysis).4.2.6 In the original analysis undertaken by the Assessment Group, the transition from stroke to non-fatal MI was excluded to avoid an inconsistency in the sponsor’s model, in which the utility applied to people experiencing an MI (after a stroke) was higher in comparison with the utility in those who did not have an MI. After consultee comments on this issue, the occurrence of non-fatal MI in this group of patients was reconsidered and, in order to explore the effect of including this transition, a recalculation was performed using aNICE Technology Appraisal 90 14relative utility decrement and a fixed additional cost in the first year after an MI. This re-analysis resulted in less favourable estimates of cost effectiveness for MR dipyridamole in combination with aspirin, and more favourable estimates for clopidogrel than the initial model. However, in all four of the scenarios presented (lifetime treatment or 2 years’ treatment, RR of non-vascular death included or excluded), either the treatment with clopidogrel was dominated or the incremental cost-effectiveness ratio (ICER) remained greater than £40,000 per QALY in people who have had an ischaemic stroke. 4.2.7 For people with MI or with PAD, the cost per additional QALY for treatment with clopidogrel was between £31,000 and £36,000 compared with aspirin, depending on assumptions made about the annual cost of PAD (lifetime analysis, excluding RR for non-vascular death). For the 2-year treatment duration, the cost per additional QALY was £17,081 for people who have had an MI and £20,733 for people who have PAD (excluding RR for non-vascular death). When effects on non-vascular death were included in the lifetime treatment analysis, the cost per QALY for clopidogrel was £94,446 (people with MI) or clopidogrel was dominated (people with PAD). For 2-year treatment duration (including the RR for non-vascular death), the cost per additional QALY was £21,448 for the MI group and £31,300 for the PAD group. 4.2.8 In order to assess the cost effectiveness of MR dipyridamole (alone) and clopidogrel in aspirin-intolerant people who have had an ischaemic stroke the Assessment Group carried out an additional analysis. The baseline risk of events in this model was estimated using data from a prospective, population-based register of stroke cases. It was assumed that the probabilities derived from this register represented the risk of further events while taking aspirin on the basis that it is considered standard therapy after a stroke and it would be reasonable to expect that most of the people included in the register would be taking aspirin. In order to derive a ‘no antiplatelet therapy’ baseline, the risk of each event was adjusted by the corresponding relative risk for placebo versus aspirin from the ESPS-2 study. The cost effectiveness of MR dipyridamole
|
|
|
|
Pneumonia duration of treatment |
Q |
|
|
|
Contrast CT findings in Parkinson disease |
Q |
|
|
|
WOF can cause osteoporosis Hypoparathyroidism RA Prolactinoma Sarcoidosis Ciclosporin therapy
|
QThese medications include, but are not limited to, aromatase inhibitors, gonadotropin-releasing hormone agonists, thyroid replacement therapy, antiepileptics, antidepressants, antipsychotics, lithium, gastric acid lowering agents, thiazolidinediones, loop diuretics, heparins and warfarin, vitamin A and cyclosporine. |
|
|
|
Pathophysiology of OP |
Osteoporosis is a progressive metabolic bone disease that decreases bone density (bone mass per unit volume), with deterioration of bone structure. Skeletal weakness leads to fractures with minor or inapparent trauma, particularly in the thoracic and lumbar spine, wrist, and hip (called fragility fractures).
Diagnosis is by dual-energy x-ray absorptiometry (DXA scan) or by confirmation of a fragility fracture.
Prevention and treatment involve risk factor modification, calcium and vitamin D supplements, exercises to maximize bone and muscle strength, improve balance, and minimize the risk of falls, and drug therapy to preserve bone mass or stimulate new bone formation.
Pathophysiology Bone is continually being formed and resorbed. Normally, bone formation and resorption are closely balanced.
Osteoblasts (cells that make the organic matrix of bone and then mineralize bone) and osteoclasts (cells that resorb bone) are regulated by parathyroid hormone (PTH), calcitonin, estrogen, vitamin D, various cytokines, and other local factors such as prostaglandins.
Peak bone mass in men and women occurs around age 30. Blacks reach higher peak bone mass than whites and Asians, whereas Hispanics have intermediate values. Men have higher bone mass than women. After achieving peak, bone mass plateaus for about 10 yr, during which time bone formation approximately equals bone resorption. After this, bone loss occurs at a rate of about 0.3 to 0.5%/yr. Beginning with menopause, bone loss accelerates in women to about 3 to 5%/yr for about 5 to 7 yr and then the rate of loss decelerates. Osteoporotic bone loss affects cortical and trabecular (cancellous) bone. Cortical thickness and the number and size of trabeculae decrease, resulting in increased porosity. Trabeculae may be disrupted or entirely absent. Trabecular bone loss occurs more rapidly than cortical bone loss because trabecular bone is more porous and bone turnover is higher. However, loss of both types contributes to skeletal fragility. Fragility fractures A fragility fracture occurs after less trauma than might be expected to fracture a normal bone. Falls from a standing height or less, including falls out of bed, are typically considered fragility fractures. The most common sites for fragility fractures are the following: Distal radius Spine (vertebral compression fractures—the most common osteoporosis-related fracture) Femoral neck Greater trochanter Other sites may include the proximal humerus and pelvis. Fractures at sites such as the nose, ribs, clavicle, and metatarsals are not considered osteoporosis-related fractures. |
|
|
|
Classification |
Classification Osteoporosis can develop as a primary disorder or secondarily due to some other factor. The sites of fracture are similar in primary and secondary osteoporosis. Primary osteoporosis More than 95% of osteoporosis in women and about 80% in men is primary. Most cases occur in postmenopausal women and older men. Gonadal insufficiency is an important factor in both men and women. Other factors that may accelerate bone loss in patients with primary osteoporosis include decreased calcium intake, low vitamin D levels, certain drugs, and hyperparathyroidism. Some patients have an inadequate intake of calcium during the bone growth years of adolescence and thus never achieve peak bone mass. The major mechanism of bone loss is increased bone resorption, resulting in decreased bone mass and microarchitectural deterioration, but sometimes bone formation is impaired. The mechanisms of bone loss may involve the following: Local changes in the production of bone-resorbing cytokines, such as increases in cytokines that stimulate bone resorption Impaired formation response during bone remodeling (probably caused by age-related decline in the number and activity of osteoblasts) Other factors such as a decline in local and systemic growth factors Fragility fractures rarely occur in children, adolescents, premenopausal women, or men < 50 yr with normal gonadal function and no detectable secondary cause, even in those with low bone mass (low Z-scores on dual-energy x-ray absorptiometry [DXA]). Such uncommon cases are considered idiopathic osteoporosis. |
Secondary osteoporosis accounts for < 5% of osteoporosis in women and about 20% in men. They may also further accelerate bone loss and increase fracture risk in patients with primary osteoporosis. Patients with chronic kidney disease may have several reasons for low bone mass, including secondary hyperparathyroidism, elevated serum phosphate, calcitriol deficiency, abnormalities of serum calcium and vitamin D, osteomalacia, and low-turnover bone disorders (adynamic bone disease). |
|
|
What is an osteoporotic compression fractures |
Pictures |
|
|
|
Q Mx of acute exacerbation of COPD ABG is a must 💡 Target saturation is 88-92% High concentrate oxygen should be used Type 2 respiratory failure with PCO2 >6kPa and pH less than 7.35 is an indication for NIV detail hx on premorbid personality is inidicated
|
Oxygen is a treatment for hypoxaemia, not breathlessness. Oxygen has not been proven to have any consistent effect on the sensation of breathlessness in non-hypoxaemic patients. ✓ The essence of this guideline can be summarised simply as a requirement for oxygen to be prescribed according to a target saturation range and for those who administer oxygen therapy to monitor the patient and keep within the target saturation range. ✓ The guideline recommends aiming to achieve normal or near-normal oxygen saturation for all acutely ill patients apart from those at risk of hypercapnic respiratory failure or those receiving terminal palliative care. Assessing patients For critically ill patients, high concentration oxygen should be administered immediately (table 1 and figure 1) and this should be recorded afterwards in the patient's health record.Clinicians must bear in mind that supplemental oxygen is given to improve oxygenation, but it does not treat the underlying causes of hypoxaemia which must be diagnosed and treated as a matter of urgency.The oxygen saturation should be checked by pulse oximetry in all breathless and acutely ill patients, ‘the fifth vital sign’ (supplemented by blood gases when necessary), and the inspired oxygen concentration should be recorded on the observation chart with the oximetry result. (The other vital signs are pulse rate, blood pressure, temperature and respiratory rate). Pulse oximetry must be available in all locations where emergency oxygen is used. Clinical assessment is recommended if the saturation falls by ≥3% or below the target range for the patient. All critically ill patients outside of a critical care area (e.g. intensive care unit (ICU), high dependency unit (HDU), respiratory HDU), should be assessed and monitored using a recognised physiological track and trigger system such as the National Early Warning Score (NEWS). Target Oxygen prescription Oxygen should be prescribed to achieve a target saturation of 94–98% for most acutely ill patients or 88–92% or patient-specific target range for those at risk of hypercapnic respiratory failure (tables 1⇓⇓–4). Best practice is to prescribe a target range for all hospital patients at the time of admission so that appropriate oxygen therapy can be started in the event of unexpected clinical deterioration with hypoxaemia and also to ensure that the oximetry section of the early warning score (EWS) can be scored appropriately.The target saturation should be written (or ringed) on the drug chart or entered in an electronic prescribing system (guidance on figure 1).Oxygen administrationOxygen should be administered by staff who are trained in oxygen administration.These staff should use appropriate devices and flow rates in order to achieve the target saturation range (figure 2).Staff should be trained in the use of a range of different oxygen delivery devices to ensure oxygen is delivered safely. Monitoring and maintenance of target saturation Oxygen saturation and delivery system (including flow rate) should be recorded on the patient's monitoring chart.Oxygen delivery devices and flow rates should be adjusted to keep the oxygen saturation in the target range. Prompt clinical assessment is required if oxygen therapy needs to be initiated or increased due to a falling saturation level.Oxygen should be prescribed and a signature should be entered on the drug chart on each drug round. Weaning and discontinuation of oxygen therapy Oxygen should be reduced in stable patients with satisfactory oxygen saturation.Oxygen should be discontinued once the patient can maintain saturation within or above the target range breathing air but the prescription for a target range should be left in place in case of future deterioration and to guide early warning scores (EWS/NEWS). |
|
|
|
DKA DIAGNOSTIC CRITERIA |
Q |
|
|
|
Q High WBC always indictate an infectionSteroid use should be doubled in a t with coexisting adrenal insufficiency
|
Even in the absence of infection, the CBC count shows an increased white blood cell (WBC) count in patients with diabetic ketoacidosis. High WBC counts (greater than 15 X 109/L) or marked left shift may suggest underlying infection.
More total leukocytes and neutrophils but fewer eosinophils was significantly correlated with DKA and DK. Leukocyte counts can add valuable information to reflect the presence of hyperglycemic crisis and acute infection. |
|
|
|
Q HyponatremiaSIADH is the commonest cause of hyonatremia3%saline (hypertonic saline)should be used to treat hyponatremia , when sodium is less than 120mmol/lPPI are a well recognized causeThyroid function tests and serum cortisol are indicated in the diagnosis of SIADHOral salt is the Rx of choice for hyponatremia due to SIADH
|
Q |
|
|
|
Digoxin toxicity |
Rapid IV infusion can cause hypotension |
|
|
|
Digoxin toxicity
cardiac glycoside toxicity continues to be a problem in the United States because of the wide use of digoxin (a preparation of digitalis) and its narrow therapeutic window.It is important to learn about the source, amount, time of ingestion, presence of any coingestant, and patient’s own comorbidities. Acute digitalis toxicity can result from unintentional, suicidal, or homicidal overdose of the digitalis preparation digoxin, or accidental ingestion of plants that contain cardiac glycosides. Chronic toxicity in patients on digoxin therapy may result from deteriorating renal function dehydration electrolyte disturbances or drug interactions.
Alterations in cardiac rate and rhythm from digitalis toxicity may reproduce almost every known mechanism of dysrhythmia. |
Signs and symptomsDigitalis toxicity produces CNS, visual, GI, and cardiac manifestations. Nausea, vomiting, and drowsiness are among the most common extracardiac manifestations.CNS symptoms of digitalis toxicity include the following:•Drowsiness•Lethargy•Fatigue•Neuralgia•Headache•Dizziness•Confusion or giddiness•Hallucinations•Seizures (rare)•Paresthesias and neuropathic painVisual aberration often is an early indication of digitalis toxicity. Yellow-green distortion is most common, but red, brown, blue, and white distortions also occur. Drug intoxication also may cause the following:•Snowy vision•Photophobia•Photopsia•Decreased visual acuity•Yellow halos around lights (xanthopsia)•Transient amblyopia or scotomataGI symptoms in acute or chronic toxicity include the following:•Anorexia•Weight loss•Failure to thrive (in pediatric patients)•Nausea•Vomiting•Abdominal pain•Diarrhea•Mesenteric ischemia (a rare complication of rapid IV infusion)Cardiac symptomsCardiac symptoms include the following:•Palpitations•Shortness of breath•Syncope•Swelling of lower extremities•Bradycardia•Hypotension•DyspneaSee Clinical Presentation for more detail.DiagnosisStudies in patients with possible digitalis toxicity include the following:•Serum digoxin level•Electrolytes•Renal function studies•ECGSerum digoxin level•Therapeutic levels are 0.6-1.3 to 2.6 ng/mL•Levels associated with toxicity overlap between therapeutic and toxic ranges•False-negative assay results may occur with acute ingestion of nondigoxin cardiac glycosides (eg, herbal compunds, such as foxglove or oleander)•Levels determined less than 6-8 hours after an acute ingestion do not necessarily predict toxicity.•The best way to guide therapy is to follow the digoxin level and correlate it with serum potassium concentrations and the patient's clinical and ECG findings. Electrolytes•In acute toxicity, hyperkalemia is common•Chronic toxicity is often accompanied by hypokalemia and hypomagnesemia Electrocardiography•Digoxin toxicity may cause almost any dysrhythmia•Classically, dysrhythmias associated with increased automaticity and decreased AV conduction occur•Sinus bradycardia and AV conduction blocks are the most common ECG changes in the pediatric population, while ventricular ectopy is more common in adults•Nonparoxysmal atrial tachycardia with heart block and bidirectional ventricular tachycardia are particularly characteristic of severe digitalis toxicity
See Workup for more detail.ManagementSupportive care of digitalis toxicity includes the following:•Hydration with IV fluids•Oxygenation and support of ventilatory function•Discontinuation of the drug, and, sometimes, the correction of electrolyte imbalancesGI decontamination•Activated charcoal is indicated for acute overdose or accidental ingestion•Binding resins (eg, cholestyramine) may bind enterohepatically-recycled digoxinTreatment of electrolyte imbalance•For hyperkalemia, use insulin plus glucose, and sodium bicarbonate if the patient is acidotic•Treatment with digoxin Fab fragments is indicated for a K+ level greater than 5 mEq/L•Hemodialysis may be necessary for uncontrolled hyperkalemia•Correct hypokalemia (usually in chronic intoxication)•Concomitant hypomagnesemia may result in refractory hypokalemiaDigoxin immune FabDigoxin immune Fab is considered the first-line treatment for significant dysrhythmias from digitalis toxicity.
Other indications for its use, in the absence of specific contraindications, include the following:•Ingestion of massive quantities of digitalis (in children, 4 mg or 0.1 mg/kg; in adults, 10 mg)•Serum digoxin level greater than 10 ng/mL in adults at steady state (ie, 6-8 hours after acute ingestion or at baseline in chronic toxicity)•Hyperkalemia (serum potassium level greater than 5 mEq/L)•Altered mental status attributed to digoxin toxicity•Rapidly progressive signs and symptoms of toxicity cardiac glycoside toxicity continues to be a problem in the United States because of the wide use of digoxin (a preparation of digitalis) and its narrow therapeutic window.It is important to learn about the source, amount, time of ingestion, presence of any coingestant, and patient’s own comorbidities. Acute digitalis toxicity can result from unintentional, suicidal, or homicidal overdose of the digitalis preparation digoxin, or accidental ingestion of plants that contain cardiac glycosides. Chronic toxicity in patients on digoxin therapy may result from deteriorating renal function, dehydration, electrolyte disturbances, or drug interactions. Alterations in cardiac rate and rhythm from digitalis toxicity may reproduce almost every known mechanism of dysrhythmia.ive inotropic effect of digitalis has the following 2 components:•Direct inhibition of membrane-bound sodium- and potassium-activated adenosine triphosphatase (Na+/K+ -ATPase), which leads to an increase in the intracellular concentration of calcium ([Ca2+]i)•Associated increase in a slow inward calcium current (iCa) during the action potential (AP); this current is the result of movement of calcium into the cell, and it contributes to the plateau of the APDigitalis glycosides bind specifically to Na+/K+ -ATPase, inhibit its enzymatic activity, and impair active transport of extruding sodium and transport of potassium into the fibers (3:2 ratio). As a result, intracellular sodium ([Na+]i) gradually increases, and a gradual, small decrease in intracellular potassium ([K+]i) occurs.Cardiac fiber calcium [Ca2+]i is exchanged for extracellular sodium (3:1 ratio) by a transport system that is driven by the concentration gradient for these ions and the transmembrane potential. Increase in [Na+]i is related crucially to the positive inotropic effect of digitalis.In addition, by a mechanism that is not defined clearly, the increase in [Ca2+]i increases the peak magnitude of iCa; this change parallels the positive inotropic action. The change in iCa is a consequence of the increase in [Ca2+]i and not of the increase in [Na+]i. Thus, more calcium is delivered during the plateau of each AP to activate each contraction.A fall in intracellular pH accompanies the digoxin-induced increase in [Ca2+] i, which leads to activation of a sodium/hydrogen exchange pump. This results in extrusion of hydrogen, an increase in [Na+]i, and greater inotropy.The mechanism described assumes that Na+/K+ -ATPase is the pharmacologic receptor for digitalis and that when digitalis binds to these enzymes, it induces a conformational change that decreases active transport of sodium. Digitalis apparently binds to ATPase in a specific and saturable manner, producing a conformational change of the enzyme such that the binding site for digitalis probably is on the external surface of the membrane. Furthermore, the magnitude of the inotropic effect of digitalis is proportional to degree of inhibition of the enzyme.Digitalis, in therapeutic concentrations, exerts no effect on the contractile proteins or on the interactions between them.Electrophysiologic effectsThe electrophysiological effects of cardiac glycosides include the following[8] :•Decreased resting potential (RP) or maximal diastolic potential (MDP), which slows the rate of phase-0 depolarization and conduction velocity•Decrease in action potential duration (APD), which results in increased responsiveness of fibers to electrical stimuli•Enhancement of automaticity, which results from an increase in the rate of phase 4 depolarization and from delayed after-depolarizationIn general, cardiac glycosides slow conduction and increase the refractory period in specialized cardiac conducting tissue by stimulating vagal tone. Digitalis has parasympathetic properties, which include hypersensitization of carotid sinus baroreceptors and stimulation of central vagal nuclei.Digoxin also appears to have variable effects on sympathetic tone, depending on the specific cardiac tissue involved.Dosage and toxicityThe therapeutic daily dose of digoxin ranges from 5-15 mcg/kg. The absorption of digoxin tablets is 70-80%; its bioavailability is 95%. The kidney excretes 60-80% of the digoxin dose unchanged.The onset of action after oral (PO) administration occurs in 30-120 minutes; the onset of action with intravenous (IV) administration occurs in 5-30 minutes. The peak effect with PO dosing is 2-6 hours, and that with IV dosing is 5-30 minutes. Only 1% of the total amount of digoxin in the body is in the serum; of that amount, approximately 25% is protein bound.Digoxin has a large volume of distribution, being 6-10 L/kg in adults, 10 L/kg in neonates, and as much as 16L/kg in infants and toddlers. At therapeutic levels, the elimination half-life is 36 hours. The lethal dose of digoxin is considered to be 20-50 times the maintenance dose taken at once. In healthy adults, a dose of less than 5 mg seldom causes severe toxicity, but a dose of more than 10 mg is almost always fatal. However, plasma concentration does not always correlate with the risk of toxicity. [9]DysrhythmiasAlterations in cardiac rate and rhythm from digitalis toxicity may simulate almost every known type of dysrhythmia. Although no dysrhythmia is pathognomonic for digoxin toxicity, toxicity should be suspected when evidence of increased automaticity and depressed conduction is noted. Underlying these dysrhythmias is a complex influence of digitalis on the electrophysiologic properties of the heart through the means already discussed, as well as via the cumulative results of the direct, vagotonic, and antiadrenergic actions of digitalis.The effects of digoxin vary with the dose and differ depending on the type of cardiac tissue involved. The atria and ventricles exhibit increased automaticity and excitability, resulting in extrasystoles and tachydysrhythmias. Conduction velocity is reduced in myocardial and nodal tissue, resulting in increased PR interval and AV block accompanied by a decrease in the QT interval.In addition to these effects, the direct effect of digitalis on repolarization often is reflected in the electrocardiogram (ECG) by ST segment and T-wave forces opposite in direction to the major QRS forces. The initial electrophysiologic manifestation of digitalis effects and toxicity usually is mediated by increased vagal tone.Early in acute intoxication, depression of sinoatrial (SA) or AV nodal function may be reversed by atropine. Subsequent manifestations are the result of direct and vagomimetic actions of the drug on the heart and are not reversed by atropine.Ectopic rhythms are due to enhanced automaticity, reentry, or both, and may include the following:•Nonparoxysmal junctional tachycardia•Extrasystole•Premature ventricular contractions•Ventricular flutter and fibrillation•Atrial flutter and fibrillation•Bidirectional ventricular tachycardiaBidirectional ventricular tachycardia is particularly characteristic of severe digitalis toxicity and results from alterations in intraventricular conduction, junctional tachycardia with aberrant intraventricular conduction, or, on rare occasions, alternating ventricular pacemakers.The following features may also be seen:•Depression of the atrial pacemakers, resulting in SA arrest•SA block•AV block•Sinus exit block resulting from depression of normal conduction•Nonparoxysmal atrial tachycardia with blockWhen conduction and the normal pacemaker are both depressed, ectopic pacemakers may take over, producing atrial tachycardia with AV block and nonparoxysmal automatic AV junctional tachycardia. Indeed, AV junctional blocks of varying degrees, alone or with increased ventricular automaticity, are the most common manifestations of digoxin toxicity, occurring in 30-40% of cases. AV dissociation may result from suppression of the dominant pacemaker with escape of a subsidiary pacemaker or inappropriate acceleration of a ventricular pacemaker.Arrhythmias can cause inadequate tissue perfusion, with resultant central nervous system (CNS) and renal complications, such as the following:•Hypoxic seizures•Encephalopathies•Loss of vasoregulation•Acute tubular necrosisHyperkalemia is the major electrolytic complication in acute, massive digoxin poisoning. In pediatric patients, hyperkalemia can be a complication of acute toxicity.EtiologyClinical digoxin toxicity represents a complex interaction between digoxin and various electrolyte and renal abnormalities. A patient with normal digoxin levels (0.5-2 ng/mL) but renal insufficiency or severe hypokalemia may have more serious cardiotoxicity than a patient with high digoxin levels and no renal or electrolyte disturbances.Acute overdose or accidental exposure to plants containing cardiac glycosides may cause acute toxicity. Deteriorating renal function, dehydration, electrolyte disturbances, or drug interactions usually precipitate chronic toxicity.The most common precipitating cause of digitalis intoxication is depletion of potassium stores, which occurs often in patients with heart failure as a result of diuretic therapy and secondary hyperaldosteronism. Dosing errors, especially in infants receiving parenteral digoxin, is a frequent cause of digoxin toxicity and is usually associated with high mortality.Toxicity may also occur via increased bioavailability. Bioavailability varies depending on the drug formulation. For example, Lanoxin has 25% less bioavailability than Lanoxicaps. Certain antibiotics that suppress intestinal flora may increase absorption of digoxin.Acute, nontherapeutic overdose—unintentional, suicidal, or homicidal—can cause toxicity. Other causes of digitalis toxicity include the following:•Advanced age•Myocardial infarction or ischemia•Hypothyroidism•Hypercalcemia•Renal insufficiency [10]•Hyperthyroidism•Hypoxemia•Alkalosis•Acidosis - Depresses the Na+/K+ATPase pump and may cause digoxin toxicity•Myocardial diseaseBoth acidosis and myocardial ischemia suppress the Na+/K+ ATPase pump. In addition, myocardial ischemia independently alters myocardial automaticity. Hypothyroid patients are prone to digoxin toxicity secondary to decreased renal excretion and a smaller volume of distribution.ElectrolytesHypomagnesemia, hypercalcemia, hypernatremia, hyperkalemia, and hypokalemia can aggravate toxicity. [11]Hypokalemia is usually observed with chronic toxicity or in patients taking diuretics; it reduces the rate of Na+/K+ATPase pump turnover and exacerbates pump inhibition due to digitalis.Hyperkalemia is the usual electrolyte abnormality precipitated by digoxin toxicity, primarily in the acute setting. Hyperkalemia may be associated with acute renal failure that subsequently precipitates digoxin toxicity. Chronic digoxin toxicity does not usually cause hyperkalemia. In pediatric patients, hyperkalemia is usually a complication of acute toxicity rather than a cause; however, preexisting hyperkalemia increases the risk of morbidity and mortality.MedicationsSome medications directly increase digoxin plasma levels; other medications alter renal excretion or induce electrolyte abnormalities. [12] Drugs that have been reported to cause digoxin toxicity include the following:•Amiloride - May reduce the inotropic response to digoxin•Amiodarone - Reduces renal and nonrenal clearance of digoxin and may have additive effects on the heart rate•Benzodiazepines (eg, alprazolam, diazepam) - Have been associated with isolated reports of digoxin toxicity•Beta-blockers (eg, propranolol, metoprolol, atenolol) - May have additive effects on the heart rate; carvedilol may increase digoxin blood levels in addition to potentiating its effects on the heart rate•Calcium channel blockers - Diltiazem and verapamil increase serum digoxin levels; not all calcium channel blockers share this effect•Cyclosporine - May increase digoxin levels, possibly due to reduced renal excretion•Erythromycin, clarithromycin, and tetracyclines - May increase digoxin levels•Propafenone - Increases digoxin level; effects are variable.•Quinidine - Increases digoxin level substantially but clinical effect is variable; related drugs, such as hydroxychloroquine and quinine, may also affect levels.•Propylthiouracil - May increase digoxin levels by reducing thyroid hormone levels•Indomethacin•Spironolactone - May interfere with digoxin assays, may directly increase digoxin levels, and may alter renal excretion•Hydrochlorothiazide•Furosemide and other loop diuretics•Triamterene•Amphotericin B - May precipitate hypokalemia and subsequent digoxin toxicity•Succinylcholine - Increased risk of dysrhythmias has been reported•Herb/nutraceutical - Ephedra increases the risk of cardiac stimulation; natural licorice causes sodium and water retention and increases potassium lossSexual and age-related differences in incidence. Older individuals with multiple comorbid conditions have a lower digitalis tolerance than do younger individuals with few or no comorbid conditions.Manifestations of digitalis toxicity vary depending on age. For instance, ventricular ectopy is most prevalent in older patients; conduction defects and supraventricular ectopic rhythms are most prevalent in younger patients. PrognosisPrognosis in digitalis toxicity worsens with increasing age and associated comorbid conditions. In general, older people have a worse outcome than other adults, who, in turn, have a worse outcome than children. Morbidity and mortality rates increase if the patient has a new dysrhythmia, advanced AV block, or other significant ECG abnormality.The lethal dose of most glycosides is approximately 5-10 times the minimal effective dose and only about twice the dose that leads to minor toxic manifestations. Morbidity is usually 4.6-10%; however, morbidity is 50% if the digoxin level is greater than 6 ng/mL.
|
|
|
|
Management of hypertensive emergencies |
Hypertensive emergencies encompass a spectrum of clinical presentations in which uncontrolled blood pressures (BPs) lead to progressive or impending end-organ dysfunction. In these conditions, the BP should be lowered aggressively over minutes to hours.
Neurologic end-organ damage due to uncontrolled BP may include hypertensive encephalopathy, cerebral vascular accident/cerebral infarction, subarachnoid hemorrhage, and/or intracranial hemorrhage. Cardiovascular end-organ damage may include myocardial ischemia/infarction, acute left ventricular dysfunction, acute pulmonary edema, and/or aortic dissection.
Other organ systems may also be affected by uncontrolled hypertension, which may lead to acute renal failure/insufficiency, retinopathy, eclampsia, or microangiopathic hemolytic anemia. With the advent of antihypertensive agents, the incidence of hypertensive emergencies in the United States has declined from 7% to approximately 1% of patients with hypertension.
In addition, the 1-year survival rate associated with this condition has increased from only 20% (prior to 1950) to a survival rate of more than 90% with appropriate medical treatment. History and physical examination The history and the physical examination determine the nature, severity, and management of the hypertensive event. The history should focus on the presence of end-organ dysfunction, the circumstances surrounding the hypertension, and any identifiable etiology.
The most common clinical presentations of hypertensive emergencies are cerebral infarction (24.5%), pulmonary edema (22.5%), hypertensive encephalopathy (16.3%), and congestive heart failure (12%).
Other clinical presentations associated with hypertensive emergencies include intracranial hemorrhage, aortic dissection, and eclampsia, as well as acute myocardial infarction, and retinal and renal involvement. In pregnant patients, acute hypertensive crisis usually results from underlying hypertensive disease or severe preeclampsia and can lead to maternal stroke, cardiopulmonary decompensation, fetal decompensation caused by reduced uterine perfusion, abruption, and stillbirth. Preeclampsia may also complicated by pulmonary edema. [9]The duration and severity of the patient’s preexisting hypertension (including the degree of BP control) should be evaluated, as well as the patient's medication history. Details of antihypertensive drug therapy and compliance, intake of over-the-counter (OTC) preparations such as sympathomimetic agents, and use of illicit drugs such as cocaine are important elements of the medication history. In addition, it is important to elicit information about the presence of previous end-organ dysfunction, particularly renal and cerebrovascular disease, and any other medical problems (eg, thyroid disease, Cushing disease, systemic lupus). In female patients, determine the date of their last menstrual period.
Patients may complain of specific symptoms that suggest end-organ dysfunction may be present.
Chest pain may indicate myocardial ischemia or infarction, back pain may denote aortic dissection; and dyspnea may suggest pulmonary edema or congestive heart failure.
The presence of neurologic symptoms may include seizures, visual disturbances, and altered level of consciousness and may be indicative of hypertensive encephalopathy.The physical examination should assess whether end-organ dysfunction is present. BP should not only be measured in both the supine position and the standing position (assess volume depletion), but it should also be measured in both arms (a significant difference may suggest aortic dissection).
The presence of new retinal hemorrhages, exudates, or papilledema suggests a hypertensive emergency.
Evaluate for the presence of heart failure, which may be indicated by jugular venous distention, crackles on auscultation, and peripheral edema.
Central nervous system (CNS) findings may include changes in the patient's level of consciousness and visual fields, and/or the presence of focal neurologic signs. Abdominal masses or bruits may be noted.
Evaluation of uncontrolled hypertension Obtain electrolyte levels, as well as measurements of blood urea nitrogen (BUN) and creatinine levels to evaluate for renal impairment.
A dipstick urinalysis to detect hematuria or proteinuria and microscopic urinalysis to detect red blood cells (RBCs) or RBC casts should also be performed
A complete blood cell (CBC) count and peripheral blood smear should be obtained to exclude microangiopathic anemia, and a toxicology screen, pregnancy test, and endocrine testing may be obtained, as needed.
Imaging should be directed by the clinical presentation. If there is clinical evidence of pulmonary edema or the patient has chest pain, chest radiography and electrocardiography are indicated.
Patients with neurologic signs should be evaluated with a head computed tomography scan initially, with more advanced imaging studies determined by the clinical presentation.
Malignant hypertension Malignant hypertension and accelerated hypertension are both hypertensive emergencies (ie, systolic BP [SBP] >180 mm Hg or diastolic BP [DBP] >120 mm Hg, and acute target organ damage with similar outcomes and therapies.
Malignant hypertension may or may not be associated with clinical conditions present in hypertensive urgency (ie, SBP >180 mm Hg or DBP >120 mm Hg, but no evidence of acute target organ damage [10, 11] ; thus, hypertensive urgency occurs in the absence of acute end-organ damage, whereas hypertensive emergencies include the presence of acute end-organ damage [11, 12] ).
A patient with malignant hypertension always has retinal papilledema (as seen in the image below), [13] as well as flame-shaped hemorrhages and exudates.
Other clinical features of malignant hypertension may include encephalopathy, confusion, left ventricular failure, intravascular coagulation, and impaired renal function, with hematuria and weight loss.
The pathologic hallmark of malignant hypertension is fibrinoid necrosis of the arterioles, which occurs systemically, but specifically in the kidneys. These patients develop fatal complications if untreated, and more than 90% will not survive beyond 1-2 years.
Papilloedema Note the swelling of the optic disc, with blurred margins.
Management of Hypertensive Emergencies Approximately 3%-45% of adult patients have at least one incident of increased blood pressure (BP) during their stay in the emergency department (ED). The fundamental principle in determining the necessary ED care of the hypertensive patient is the presence or absence of end-organ dysfunction. Many patients present to the ED with elevated BPs; however, only a small proportion of patients will require emergency treatment. An important point to remember in the management of the patient with any degree of BP elevation is to "treat the patient and not the number."
The primary goal of the emergency physician is to determine which patients with acute hypertension are exhibiting symptoms of end-organ damage and require immediate intravenous (IV) parenteral therapy.
In contrast, patients presenting with acutely elevated BP (systolic BP [SBP] >200 mm Hg or diastolic BP [DBP] >120 mm Hg) without symptoms and whose BP stays significantly elevated to this level on discharge should have initiation of medical therapy and close follow-up in the outpatient setting, with BP reduction over hours or days.
Thus, optimal control of hypertensive situations balances the benefits of immediate decreases in BP against the risk of a significant decrease in target organ perfusion.
Acutely lowering BP in the ED for clinical situations other than those listed below is controversial and generally should be avoided. The long-term prognosis for patients with hypertensive emergencies or urgencies is not good.
Pharmacotherapy Optimal pharmacotherapy is dependent upon the specific organ at risk (see the individual sections below). In patients presenting with hypertensive emergencies, antihypertensive drug therapy has been shown to be effective in acutely decreasing BP.
Sodium nitroprusside is a commonly used medication. It is a short-acting agent, and the BP response can be titrated from minute to minute. However, patients must have constant monitoring in an ICU. The potential exists for thiocyanate and cyanide toxicity with prolonged use or if the patient has renal or hepatic failure.
Labetalol, an alpha- and beta-blocking agent, has proven to be quite beneficial in the treatment of patients with hypertensive emergencies. Labetalol is particularly preferred in patients with acute dissection and those with end-stage renal disease. Boluses of 10-20 mg may be administered, or the drug may be infused at 1 mg/min until the desired BP is obtained. Once an adequate BP level is obtained, oral hypertensive therapy should be initiated, and patients are gradually weaned from parenteral agents.
Fenoldopam, a peripheral dopamine-1-receptor agonist is given as initial IV dose of 0.1 µg/kg/min titrated every 15 minutes.
Clevidipine, a dihydropyridine CCB, is administered IV for rapid and precise BP reduction. It is rapidly metabolized in the blood and tissues and does not accumulate in the body. Initiate IV infusion of clevidipine at 1-2 mg/hour; titrate the dose at short intervals (ie, 90 seconds) initially by doubling the dose.As the BP approaches its goal, increase the clevidipine dose by less than double, and lengthen the time between dose adjustments to every 5-10 minutes. An approximately 1-2 mg/hour increase produces an additional 2-4 mm Hg decrease in SBP. Typically, the therapeutic response is achieved with 4-6 mg/hour, although severe hypertension may require higher doses. Most patients have received maximum doses of 16 mg/hour or less; experience is limited with short-term dosing as high as 32 mg/hour. Because of lipid load restrictions, do not exceed 1000 mL or an average of 21 mg/hour within a 24-hour period; experience is limited with use beyond 72 hours. |
Guidelines recommendationsThe 2017 American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommendations for hypertensive crises and emergencies include the following :Admit adults with a hypertensive emergency to an ICU for continuous monitoring of BP and target organ damage, as well as for parenteral administration of an appropriate medication.
For adults with a compelling condition (ie, aortic dissection, severe preeclampsia or eclampsia, or pheochromocytoma crisis), lower SBP to below 140 mm Hg during the first hour and to below 120 mm Hg in aortic dissection.
For adults without a compelling condition, reduce the SBP to a maximum of 25% within the first hour; then, if the patient is clinically stable, lower the BP to 160/100 -110 mm Hg over the next 2-6 hours, and then cautiously to normal over the following 24-48 hours.
Neurologic emergencies BP reduction is indicated in neurologic emergencies, such as hypertensive encephalopathy, acute ischemic stroke, acute intracerebral hemorrhage, and subarachnoid hemorrhage.
Hypertensive encephalopathy In hypertensive encephalopathy, the treatment guidelines are to reduce the MAP 25% over 8 hours. Labetalol, nicardipine, esmolol are the preferred medications; nitroprusside and hydralazine should be avoided.
Acute ischemic stroke For acute ischemic stroke, the preferred medications are labetalol and nicardipine.
Withhold antihypertensive medications unless the SBP is above 220 mm Hg or the DBP is over 120 mm Hg, UNLESS the patient is eligible for IV tissue plasminogen activator (tPA); then, the goal is a gradual reduction of BP with a goal SBP of less than 185 mm Hg and a DBP below 110 mm Hg before initiating thrombolitic therapy.
After initiating drug therapy but before administering tPA, the SBP should be maintained at less than 180 mm Hg and the DBP below 105 mm Hg for 24 hours.
Acute intracerebral hemorrhage For acute intracerebral hemorrhage, the preferred medications are labetalol, nicardipine, and esmolol; avoid nitroprusside and hydralazine.
The treatment is based on clinical/radiographic evidence of increased intracranial pressure (ICP). If there are signs of increased ICP, maintain the MAP just below 130 mm Hg (or SBP 26</ref>
In adults with acute intracerebral hemorrhage who present with an SBP above 220 mm Hg, continuous IV drug and close BP monitoring is reasonable to lower SBP. Note that it may be harmful to immediately lower SBP to below 140 mm Hg in adults with spontaneous intracerebral hemorrhage who present within 6 hours of the acute event and have an SBP between 150 and 220 mm Hg.
Subarachnoid hemorrhage
In subarachnoid hemorrhage, nicardipine, labetalol, and esmolol are also the preferred agents; again, nitroprusside and hydralazine should be avoided. Maintain the SBP below 160 mm Hg until the aneurysm is treated or cerebral vasospasm occurs.
Although oral nimodipine is used to prevent delayed ischemic neurologic deficits, it is NOT indicated for treating acute hypertension.
Cardiovascular emergencies Rapid BP reduction is also indicated in cardiovascular emergencies, such as aortic dissection, acute coronary syndrome, and acute heart failure.
Aortic dissection Beta blockers are the recommended antihypertensive agents in patients with hypertension and thoracic aortic disease. In aortic dissection, the preferred medications are labetalol, nicardipine, nitroprusside (with beta-blocker), esmolol, and morphine sulfate. However, avoid beta-blockers if there is aortic valvular regurgitation or suspected cardiac tamponade.For adults with aortic dissection, rapidly lower the SBP to below 120 mm Hg; the preferred agents are esmolol and labetalol. Beta blockade should precede vasodilator administration, if needed for BP control or to prevent reflex tachycardia or inotropic effect; achieve SBP up to 120 mm Hg within 20 minutes.
Maintain the SBP below 110 mm Hg, unless signs of end-organ hypoperfusion are present. The preferred treatment includes a combination of narcotic analgesics (morphine sulfate), beta blockers (labetalol, esmolol), and vasodilators (nicardipine, nitroprusside). CCBs (verapamil, diltiazem) are an alternative to beta blockers.
Acute coronary syndrome For acute coronary syndrome, beta blockers and nitroglycerin are the preferred drugs. Treatment is indicated if the SBP is above 160 mm Hg and/or the DBP is over 100 mm Hg. Reduce the BP by 20%-30% of baseline. Note that thrombolytics are contraindicated if the BP is above 185/100 mm Hg.
In addition, note that nitrates administered in the presence of phosphodiesterase type 5 (PDE-5) inhibitors may induce profound hypertension.
Acute heart failureIn acute heart failure, the preferred medications are IV nitroglycerin or sublingual nitroglycerin and IV enalaprilat. Treat with vasodilators (in addition to diuretics) for a SBP of 140 mm Hg. In adults with hypertension at an increased risk of heart failure, the optimal BP should be below 130/80 mm Hg. In hypertensive adults with reduced ejection fraction (HFrEF), prescribe guideline-directed medical therapy (GDMT) to achieve a BP below 130/80 mm Hg.
Note that nondihydropyridine CCBs are not recommended for treatment in this patient population.
In hypertensive adults with preserved ejection fraction (HFpEF) and symptoms of volume overload, prescribe diuretics to control BP. For those with persistent hypertension after management of volume overload, prescribe ACEIs or ARBs and beta blockers titrated to achieve an SBP below 130 mm Hg. |
|
|
Q Repeated nebulisation with salbutamol is indicated in severe hyperkalemia |
For several years now, it has been known that the administering of adrenergic beta antagonists, especially of the beta-2 type, induce hypokalemia as a result of the entering of potassium into the skeletal muscle cells. This fall in kalemia occurs independently from the effect of insulin, aldosterone or kidney excretion, is mediated by the beta-2 receptors and require the intervention of cAMP joined at the cell membrane and the subsequent stimulation of the Na-K-ATPase which bring the potassium into the striated muscle cell. Among the most outstanding drugs with beta-2 effect is salbutamol, which maintains the hypokalemic effect whether administered intravenously or inhaled. It has been used in cases of hyperkalemia, in both children and adults. The initially used intravenous dosage (0.5 mg) caused several side-effects, especially rapid heart beat, seen more in children. It has been recently found that the use of doses as low as 4 micrograms/kg lower the kalemia to values averaging 1.4 to 1.6 mEq/L (mmol/L); in addition, using these dosages intravenously in an average of 20 minutes, no side-effects were seen, even when administered to newborns. For the above, we considered that salbutamol, in the suggested dosages, constitutes an efficient and secure therapeutic method for the initial treatment of severe hyperkalemic patients. |
|
|
|
Lactulose is an established remedy for hepatic encephalopathy and shows efficacy for chronic renal insufficiency, reducing volume overload, uremia and hyperkalemia.
Potentially lactulose could also be used for non-diuretic treatment of congestive heart failure.
However, use of lactulose is limited by diarrhea and flatulence.
Chronic lactulose administration might be tolerable if it was accomplished by nocturnal infusion through a percutaneous duodenostomy tube, also placing a rectal foley each night following a clearing enema so that large volumes of liquid stool could be passed while patients sleep.
Each morning the duodenostomy would be clamped and the foley removed.
For acute patients without duodenostomies, a temporary dobhoff feeding tube with accompanying rectal foley could be employed.
Patients who did not want a rectal foley could elect to have a permanent colostomy.
Clinical trials could establish the relationship between lactulose infusion and clearance of water, salt, potassium, hydrogen, urea and other wastes, and compare efficacy, cost and tolerability with that of peritoneal dialysis and ultrafiltration.
Lactulose could potentially allow inexpensive home-based therapy for hepatic encephalopathy, chronic renal failure and congestive heart failure, and might be life-saving in countries where renal replacement in any form is currently unavailable. |
Q Rx should be strated promptly in a pt with suggestive ECg changes without waiting for electrolyte levels
|
This site is intended for healthcare professionalsMedscape LogoDrugs & Diseases > NephrologyHyperkalemia MedicationUpdated: Jun 20, 2018 Author: Eleanor Lederer, MD, FASN; Chief Editor: Vecihi Batuman, MD, FASN more...Share FeedbackSECTIONSMedication SummaryThe goals of pharmacotherapy are to reduce potassium levels and morbidity and to prevent complications. Calcium protects the myocardium from the deleterious effects of hyperkalemia. Beta-adrenergic agents, insulin, and loop diuretics stimulate cellular uptake of potassium, lowering the serum potassium level.Calcium saltsClass SummaryCalcium antagonizes the cardiotoxicity of hyperkalemia by stabilizing the cardiac cell membrane against undesirable depolarization. Onset of effect is rapid (≤ 15 minutes) but relatively short-lived. These agents are the first-line treatment for severe hyperkalemia (ie, >7 mEq/L), when the electrocardiogram (ECG) shows significant abnormalities (eg, widening of QRS interval, loss of P wave, or cardiac arrhythmias). Calcium usually is not indicated when the ECG shows only peaked T waves.Calcium has no effect on the serum level of potassium. For that reason, administration of calcium should be accompanied by the use of other therapies that actually help lower serum potassium levels.Calcium chloride contains about 3 times more elemental calcium than an equal volume of calcium gluconate: 1 g of calcium chloride has 270 mg (13.5 mEq) of elemental calcium, whereas 1 g of calcium gluconate has 90 mg (4.5 mEq). Therefore, when hyperkalemia is accompanied by hemodynamic compromise, calcium chloride is preferred to calcium gluconate. Other calcium salts (eg, glubionate and gluceptate) have even less elemental calcium than calcium gluconate and generally are not recommended for therapy of hyperkalemia.Calcium gluconateView full drug informationCalcium increases the threshold potential, thus restoring the normal gradient between threshold potential and resting membrane potential, which is abnormally elevated in hyperkalemia. Onset of action is within 5 minutes, and duration of action is about 30-60 minutes. Doses should be titrated with constant monitoring of ECG changes during administration; repeat the dose if ECG changes do not normalize within 3-5 minutes.Calcium chlorideView full drug informationCalcium prevents the deleterious cardiac effects of severe hyperkalemia that may occur before the serum potassium level is corrected. Because of its irritating effects when administered parenterally, calcium chloride is generally considered a second choice, after calcium gluconate.Beta-adrenergic agonistsClass SummaryThrough activation of cyclic adenosine monophosphate (cAMP), these agonists stimulate the sodium-potassium–adenosine triphosphatase (Na+ -K+ -ATPase) pump, thereby shifting potassium into the intracellular compartment. However, these shifts in potassium occur primarily during exercise rather than at rest.Albuterol (Proventil, Ventolin, Vospire ER)View full drug informationAlbuterol is an adrenergic agonist that has an additive effect with insulin and glucose, which may in turn help shift potassium into the intracellular space. This agent lowers the serum potassium level by 0.5-1.5 mEq/L. It can be very beneficial in patients with renal failure when fluid overload is concern. Onset of action is 30 minutes; duration of action is 4-6 hours for the immediate-release product.Antidiabetics, InsulinsClass SummaryInsulin is administered with glucose to facilitate the uptake of glucose into muscle cells, bringing potassium with it, primarily by enhancing the activity of the Na+ -K+ -ATPase pump and thereby temporarily lowering serum potassium levels.Insulin regular human (Novolin R, Humulin R)View full drug informationRegular insulin stimulates cellular uptake of potassium within 20-30 minutes and lasts for 4-6 hours. The serum potassium concentration typically drops by 0.5-1.2 mEq/L. Administer glucose along with insulin to prevent hypoglycemia. Monitor blood sugar levels frequently. Although the effect is rapid, it is temporary; therefore, insulin therapy should be followed by therapy that actually enhances potassium clearance (eg, sodium polystyrene sulfonate [SPS]).Diuretics, LoopClass SummaryLoop diuretics markedly enhance renal potassium excretion and thus lower serum levels. Parenterally administered drugs have a more rapid onset of action and are preferable in emergency situations. Simultaneous administration of saline can prevent severe volume depletion.Furosemide (Lasix)View full drug informationFurosemide increases excretion of water by interfering with the chloride-binding cotransport system, which, in turn, inhibits sodium, potassium, and chloride reabsorption in the ascending loop of Henle and distal renal tubule. Furosemide has a slow onset of action (frequently 1 hour), and its effect on lowering the potassium level is inconsistent. Large doses may be needed in renal failure.Individualize the dose to the patient. For the treatment of edema, depending on the response, administer in increments of 20-40 mg, no sooner than 6-8 hours after the previous dose, until the desired diuresis occurs. When treating infants and children, give 1-2 mg/kg every 6-12 hours. If the diuretic response is not satisfactory, furosemide may be titrated in increments of 1 mg/kg (no sooner than 2 hours after the previous dose) until a satisfactory effect is achieved (up to 6 mg/kg).Oral absorption of furosemide varies from person to person. If the patient requires rapid and effective therapy, the intravenous (IV) route is preferred. Continuous infusion of furosemide (at rates as high as 40 mg/hr) is occasionally used for severe edema but rarely is required for the treatment of hyperkalemia.Bumetanide (Bumex)View full drug informationBumetanide increases excretion of water by interfering with the chloride-binding cotransport system, which, in turn, inhibits sodium, potassium, and chloride reabsorption in the ascending loop of Henle and distal renal tubule. Individualize the dose to the patient.For treatment of edema in adults, start at 0.5-1 mg IV or intramuscularly (IM); if the desired response is not achieved, administer a second or third dose at 2-3 hour intervals. Titrate to a maximum dosage of 10 mg/day. Rarely, dosages as high as 20 mg/day are used for edema in patients with renal impairment; however, they generally are not required for treatment of hyperkalemia.Ethacrynic acid (Edecrin)View full drug informationEthacrynic acid increases excretion of water by interfering with the chloride-binding cotransport system, which in turn inhibits sodium and chloride reabsorption in the ascending loop of Henle and distal renal tubule. For treatment of edema in adults, start at 0.5-1 mg/kg IV. Typically, 1 dose is all that is needed; occasionally, however, a second dose may be given after 2-4 hours. For second doses, a new injection site should be used so as to avoid possible thrombophlebitis. Single IV doses higher than 100 mg are not recommended.Potassium BindersClass SummaryPotassium binders are cationic exchange resins that enhance fecal excretion of potassium.Sodium polystyrene sulfonate (Kayexalate, Klonex, Kalexate, SPS)View full drug informationSPS exchanges sodium for potassium and binds it in the gut, primarily in the large intestine, decreasing the total body potassium level by approximately 0.5-1 mEq/L. Multiple doses are usually necessary.Onset of action ranges from 2 to 24 hours after oral administration and is even longer after rectal administration. The duration of action is 4-6 hours. Do not use SPS as a first-line therapy for severe life-threatening hyperkalemia; use it in the second stage of therapy.The US Food and Drug Administration (FDA) notes that SPS has been associated with intestinal necrosis and other serious gastrointestinal (GI) complications and advises against its use in patients who do not have normal bowel function. Concomitant use of sorbitol with sodium polystyrene sulfonate has been implicated in cases of colonic necrosis. [62]Patiromer (Veltassa)View full drug informationPatiromer sorbitex calcium is a nonabsorbed, cation exchange polymer that contains a calcium-sorbitol counterion. It increases fecal potassium excretion by binding potassium in the lumen of the GI tract. It is indicated for hyperkalemia. It should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action.Sodium zirconium cyclosilicate (Lokelma)View full drug informationPotassium binder; nonabsorbed zirconium silicate that preferentially captures potassium in exchange for hydrogen and sodium. It increases fecal potassium excretion through binding of potassium in the lumen of the GI tract; binding of potassium reduces the free potassium concentration in the GI lumen, thereby lowering serum potassium level. It is indicated for treatment of nonemergent hyperkalemia in adults.Alkalinizing AgentsClass SummaryIn patients with severe metabolic acidosis, sodium bicarbonate IV is used as a buffer that breaks down to water and carbon dioxide after binding free hydrogen ions. By increasing the pH, sodium bicarbonate promotes a temporary potassium shift from the extracellular to the intracellular environment. It also enhances the effectiveness of insulin in patients with acidemia. These agents have been successfully used in the treatment of acute overdose of slow-release oral potassium preparations.The use of sodium bicarbonate can be considered in treatment of hyperkalemia even in the absence of metabolic acidosis, though it is less likely to be effective in this context. This agent also increases sodium delivery to the kidney, which assists in potassium excretion .Sodium bicarbonateView full drug informationThe bicarbonate ion neutralizes hydrogen ions and raises urinary and blood pH. Onset of action occurs within minutes; duration of action is approximately 15-30 minutes. Monitor blood pH to avoid excess alkalosis. Use the 8.4% solution in adults and children and the 4.2% solution in children younger than 2 years. The adult dose for hyperkalemia is 50 mEq IV over 5 minutes. Consider methods of enhancing potassium removal or excretion, as appropriate.The following formula may be used to estimate the dose that should be administered for metabolic acidosis:HCO3− (mEq) = 0.5 (L/kg) × weight (kg) × (24 − serum HCO3− [mEq/L])This formula has many limitations; however, it allows the practitioner to make a rough determination of the amount of bicarbonate required and subsequently to titrate against the pH and anion gap.ElectrolytesClass SummaryMagnesium sulfate is used for hyperkalemic patients with cardiac arrhythmias from digitalis toxicity.Magnesium sulfateView full drug informationMagnesium is a cofactor in enzyme systems involved in neurochemical transmission and muscular excitability. In adults, potassium 60-180 mEq/day, magnesium 10-30 mEq/day, and phosphate 10-40 mmol/day may be necessary for optimum metabolic response. Give IV for acute suppression of torsades de pointes. Repeat doses are dependent on the continuing presence of patellar reflex and adequate respiratory function. |
|
|
Diabetic nephropathy When to stop metformin Cr value Type 4 RTA Retinopathy and nephropathy association |
CONCLUSIONS: Prevalence and risk factors for microangiopathy are similar to other studies, and the important finding is that the TC/HDL ratio was significant for DME. Microalbuminuria is a risk factor for diabetic retinopathy in type 1 diabetes mellitus patients but not for type 2. Overt nephropathy is well correlated with diabetic retinopathy.
Type 1 Progression of retinopathy and development of nephropathy each increase the risk for incidence of the other, independent of established risk factors for microvascular complications. |
|
|
|
Sepsis markers . T/F regarding markers of severe community acquired pneumonia
CURB-65 score more than 2 Multilobar involvement in CXR Hyponatremia Hypoalbuminaemia 💡 Positive blood culture 💡
|
Sepsis is a clinical syndrome of life-threatening organ dysfunction caused by a dysregulated response to infection. In septic shock, there is critical reduction in tissue perfusion; acute failure of multiple organs, including the lungs, kidneys, and liver, can occur.
Common causes in immunocompetent patients include many different species of gram-positive and gram-negative bacteria. Immunocompromised patients may have uncommon bacterial or fungal species as a cause.
Signs include fever, hypotension, oliguria, and confusion.
Diagnosis is primarily clinical combined with culture results showing infection; early recognition and treatment is critical.
Treatment is aggressive fluid resuscitation, antibiotics, surgical excision of infected or necrotic tissue and drainage of pus, and supportive care.
Sepsis represents a spectrum of disease with mortality risk ranging from moderate (eg, 10%) to substantial (eg, > 40%) depending on various pathogen and host factors along with the timeliness of recognition and provision of appropriate treatment.
Septic shock is a subset of sepsis with significantly increased mortality due to severe abnormalities of circulation and/or cellular metabolism. Septic shock involves persistent hypotension (defined as the need for vasopressors to maintain mean arterial pressure ≥ 65 mm Hg, and a serum lactate level > 18 mg/dL [2 mmol/L] despite adequate volume resuscitation [1]).The concept of the systemic inflammatory response syndrome (SIRS), defined by certain abnormalities of vital signs and laboratory results, has long been used to identify early sepsis.
However, SIRS criteria have been found to lack sensitivity and specificity for increased mortality risk, which is the main consideration for using such a conceptual model. The lack of specificity may be because the SIRS response is often adaptive rather than pathologic.
Diagnosis Clinical manifestations BP, heart rate, and oxygen monitoring CBC with differential, electrolyte panel and creatinine, lactate Invasive central venous pressure (CVP), PaO2, and central venous oxygen saturation (ScvO2) readings Cultures of blood, urine, and other potential sites of infection, including wounds in surgical patients
Sepsis is suspected when a patient with a known infection develops systemic signs of inflammation or organ dysfunction. Similarly, a patient with otherwise unexplained signs of systemic inflammation should be evaluated for infection by history, physical examination, and tests, including urinalysis and urine culture (particularly in patients who have indwelling catheters), blood cultures, and cultures of other suspect body fluids. In patients with a suspected surgical or occult cause of sepsis, ultrasonography, CT, or MRI may be required, depending on the suspected source.
Blood levels of C-reactive protein and procalcitonin are often elevated in severe sepsis and may facilitate diagnosis but they are not specific.
Ultimately, the diagnosis is clinical.
Other causes of shock (eg, hypovolemia, MI) should be ruled out via history, physical examination, ECG, and serum cardiac markers.
Even in the absence of MI, hypoperfusion caused by sepsis may result in ECG findings of ischemia including nonspecific ST-T wave abnormalities, T-wave inversions, and supraventricular and ventricular arrhythmias.
It is important to detect organ dysfunction as early as possible. A number of scoring systems have been devised, but the sequential organ failure assessment score (SOFA score) and the quick SOFA score (qSOFA) have been validated with respect to mortality risk and are relatively simple to use. The qSOFA criteria identify patients who should have further clinical and laboratory investigation (all 3 criteria must be present): Respiratory rate ≥ 22/min Altered mentation Systolic BP ≤ 100 mm Hg
The SOFA score is somewhat more robust but requires laboratory testing (see Table: Sequential Organ Failure Assessment (SOFA) Score).
Sequential Organ Failure Assessment (SOFA) ScoreiconCLINICAL CALCULATOR: Glasgow Coma Scaleicon CBC, ABGs, chest x-ray, serum electrolytes, BUN and creatinine, Pco2, and liver function are monitored.
Serum lactate levels, central venous oxygen saturation (ScvO2), or both can be done to help guide treatment.
WBC count may be decreased (< 4,000/μL) or increased (> 15,000/μL), and PMNs may be as low as 20%.
During the course of sepsis, the WBC count may increase or decrease, depending on the severity of sepsis or shock, the patient's immunologic status, and the etiology of the infection. Concurrent corticosteroid use may elevate WBC count and thus mask WBC changes due to trends in the illness.
Hyperventilation with respiratory alkalosis (low Paco2 and increased arterial pH) occurs early, in part as compensation for lactic acidemia. Serum bicarbonate is usually low, and serum and blood lactate levels increase.
As shock progresses, metabolic acidosis worsens, and blood pH decreases. Early hypoxemic respiratory failure leads to a decreased PaO2:FIO2 ratio and sometimes overt hypoxemia with Pao2< 70 mm Hg. Diffuse infiltrates may appear on the chest x-ray due to acute respiratory distress syndrome (ARDS). BUN and creatinine usually increase progressively as a result of renal insufficiency.
Bilirubin and transaminases may rise, although overt hepatic failure is uncommon in patients with normal baseline liver function.
Many patients with severe sepsis develop relative adrenal insufficiency (ie, normal or slightly elevated baseline cortisol levels that do not increase significantly in response to further stress or exogenous ACTH). Adrenal function may be tested by measuring serum cortisol at 8 AM; a level < 5 mg/dL is inadequate. Alternatively, cortisol can be measured before and after injection of 250 mcg of synthetic ACTH; a rise of < 9 mcg/dL is considered insufficient.
However, in refractory septic shock, no cortisol testing is required before starting corticosteroid therapy.
Hemodynamic measurements with a central venous or pulmonary artery catheter (see Monitoring and Testing the Critical Care Patient : Procedure) can be used when the specific type of shock is unclear or when large fluid volumes (eg, > 4 to 5 L 0.9% saline over 6 to 8 h) are needed. Bedside echocardiography in the ICU is a practical and noninvasive alternative method of hemodynamic monitoring. In septic shock, cardiac output is increased and peripheral vascular resistance is decreased, whereas in other forms of shock, cardiac output is typically decreased and peripheral resistance is increased.
Neither CVP nor pulmonary artery occlusive pressure (PAOP) is likely to be abnormal in septic shock, unlike in hypovolemic, obstructive, or cardiogenic shock.
|
|
|
|
Q
WOF drugs can be used in the symptom control of angina Atenolol Diltiazem Nicorandil Ivabradine Ranolazine
|
Medications that provide symptomatic relief but that have not been shown to affect long-term major events include nitrates (eg, nitroglycerin IV), calcium channel blockers (eg, diltiazem, verapamil), and heparin. Medications that have been convincingly shown to be capable of reducing short- or long-term adverse events are as follows: Aspirin Clopidogrel Beta-adrenergic blocking agents Lipid-lowering agents (statins) Angiotensin-converting enzyme (ACE) inhibitors Glycoprotein (GP) IIb/IIIa antagonists |
|
|
|
Copd |
. The review of the findings of the articles showed a significant relationship between COPD and cognitive impairment. The most widely studied cognitive domains are memory and attention.
Verbal memory and learning constitute the second most commonly impaired cognitive domain in patients with COPD.
The prevalence of impairment in visuospatial memory and intermediate visual memory is 26.9% and 19.2%, respectively.
We found that cognitive impairment is associated with the profile of COPD severity and its comorbidities.
The articles reviewed demonstrated that there is considerable impairment of the cognitive domains memory and attention in patients with COPD.
The hallmark of COPD is chronic airflow obstruction that has a systemic impact and a progressive evolution.( 1 ) It is an important health problem that is estimated to become the fifth leading cause of disability and the third leading cause of death worldwide by 2020.( 2 ) The prevalence of COPD in the global population is close to one percent and increases with age. Among individuals 40 years of age or older in the city of São Paulo, Brazil, its prevalence ranges from 6 to 15.8%.( 3 )The typical profile of patients with COPD includes multiple comorbidities,( 4 , 5 ) such as heart disease,( 6 ) osteoporosis,( 7 ) type 2 diabetes mellitus,( 8 ) lung cancer,( 9 ) and cognitive impairment.( 10 ) In recent years, the clinical relevance of cognitive impairment has risen,( 11 ) due to the increase in the prevalence of COPD and the growing interest in the aspects that determine functionality and treatment compliance( 12 , 13 ) among patients with the disease. |
However, our review of the literature clearly showed the existence of a relationship between COPD and cognitive impairment. That relationship appears to be determined by the severity of COPD and by its comorbidities.The most widely studied cognitive domains are memory and attention, both of which have been explored with specific assessment tools and found to be considerably impaired in patients with COPD. |
|
|
CSF PLEOCYTOSIS |
MedicineWolters Kluwer HealthCerebrospinal fluid pleocytosis in infectious and noninfectious central nervous system diseaseA retrospective cohort studyGertrud Baunbæk Egelund, MD, Gideon Ertner, MD, [...], and Christian T. Brandt, MD, DMScAdditional article informationAbstractCerebrospinal fluid (CSF) analysis is the most important tool for assessing central nervous system (CNS) disease. An elevated CSF leukocyte count rarely provides the final diagnosis, but is almost always an indicator of inflammation within the CNS.The present study investigated the variety of diseases associated with CSF pleocytosis.CSF analyses were identified through the biochemical database used in the capital region of Denmark in the period from 2003 to 2010. In patients >15 years, clinical diagnoses associated with the finding of a CSF leukocyte count >10 × 106 cells/L were obtained from discharge records and patient files.A total of 1058 CSF samples from 1054 patients were included in the analysis. The median age was 50 (interquartile range: 36–67) and 53% were male. Eighty-one different diagnoses were identified in 1058 cases with an elevated CSF leukocyte count, besides unknown causes. Infections were the most common cause of CSF pleocytosis (61.4%) followed by miscellaneous causes (12.7%), vascular (9.7%), neurodegenerative (7%), neoplastic (5%), and inflammatory conditions (4.2%). Only infections presented with leukocyte counts >10,000 × 106/L. Infections represented 82.6% of all cases with a leukocyte count >100 × 106/L whereas 56.3% of cases with at leukocyte counts <100 × 106/L were dominated by disease not related to infection.The present study may serve as a reminder to clinicians of what diseases and disease categories to suspect when patients present with CSF biochemistry indicating CNS inflammation.Keywords: central nervous system infections, central nervous system pleocytosis, diagnostics1. IntroductionAny means which will facilitate the difficult diagnosis of diseases of the central nervous system is of value, and the cerebrospinal fluid, which bathes its deepest recesses and washes the very nerve cells and fibers themselves, is in truth a mirror which reflects every change taking place in that system.[1]The value of analyzing the cerebrospinal fluid (CSF) was recognized even before these words were written almost a hundred years ago, and has not diminished since then despite major advances in diagnostics. In particular, the presence and number of leukocytes in the CSF has been a major criterion in the diagnosis of diseases of the central nervous system (CNS).[2]CSF analysis is a high-priority analysis upon suspicion of CNS disease. A variety of biomarkers indicating degenerative neurological disease are available, these are not specific and can merely serve as a guide to the clinical evaluation. Thus, clinician in the initial evaluation must rely on “basic” CSF biochemistry. An elevated CSF leukocyte count rarely provides the final diagnosis, but may indicate an inflammatory process within the CNS and is essential in guiding subsequent decision making and management. In addition to this, the level of CSF protein, CSF/blood albumin ratio, and glucose provide important information on indices of blood–brain barrier breakdown and the presence of glucose consuming cells/organisms.Recruitment of leukocytes to the CSF is mediated by the release of cytokines and chemokines that attract leukocytes and open up the endothelial barrier, allowing leukocyte chemotaxis. The number of processes, cell types, and molecules identified as being involved in cellular recruitment is increasing and occurs in a broad spectrum of diseases covering infectious, inflammatory (autoimmune), metabolic, and malignant diseases.[3]Insight into the biochemical profile of the CSF in different diseases should be useful knowledge to the clinical decision-making regarding treatment and supplemental diagnostics. To the best of our knowledge no such overview is available in the published literature. In this study, we therefore aimed to document the variety of conditions that may cause or be associated with CSF indices of neuroinflammation.2. Methods and materials2.1. Design, setting, and populationThis retrospective study is based on the collection of all CSF cell counts and additional biochemistry from 2 hospitals from 2003 to 2010 and from an additional 3 hospitals from 2008 to 2010. All hospitals were located within the Capital Region of Denmark.2.2. Inclusion criteriaCSF samples with a leukocyte count >10 × 106 cells/L from patients older than 15 years of age were included in the study.2.3. Exclusion criteriaCSF samples with an elevated CSF count due to blood contamination; resampling in the same patient within 90 days of the primary sample; samples related to a neurosurgical procedure; and samples where patient records are missing were excluded from the study.2.4. Data collection and variablesAll CNS analyses in the study period were retrieved from the biochemistry databases LABKA I and LABKA II (CSC Scandihealth, Aarhus, Denmark). Data on CSF leukocyte counts were extracted automatically from the biochemistry databases. Samples with a CSF leukocyte count <10 × 106 leukocytes/L were excluded. Three medical doctors (GBE, KLK, and CTB) reviewed all patient files for a discharge diagnosis according to the International Statistical Classification of Diseases and Related Health Problems, 10th Revision from the World Health Organization. The 3 medical doctors (GBE, KLK, and CTB) evaluated all cases to ensure that the diagnosis was in agreement with information from the patient files and discharge records. All data were then reviewed and verified by a fourth medical doctor (GE). Cases of CNS infections diagnosed with encephalitis or viral meningitis but with positive bacterial CSF culture and CSF biochemistry in accordance with the diagnosis were corrected and vice versa. If a diagnosis related to the finding of CSF leukocytosis was not registered, and no cause for this was available from the patient records we registered the diagnosis as “unknown.”CSF cell count was determined automatically. For each CSF sample proportion of neutrophils (%), protein (g/L), and glucose (mmol/L) level as well as the patients’ age and sex were recorded. Data on biochemical, microbiological, and radiological data were collected manually by the authors.2.5. Diagnosis categoriesThe diagnoses were a priori divided into 6 groups based on the proposed pathogenesis of the disease: infectious, inflammatory, neurodegenerative, neoplastic, vascular, and miscellaneous. Since infections were the most common cause of CSF leukocytosis, a subgroup analysis was performed dividing infectious causes into the following groups: acute bacterial, chronic bacterial, fungal, and viral CNS infections and inflammation secondary to an infectious focus outside the CNS (extra-CNS).2.6. StatisticsDescriptive data are reported as counts and percentage, and as medians and range. Information on missing data is provided in the tables. Predictive associations between CSF leukocyte count and a diagnosis of bacterial meningitis (BM) were analyzed using receiver operating characteristics (ROC). Youden J statistic/index (J = Sensitivity + (Specificity − 1)) was calculated in order to select a cut-off for maximizing classification accuracy.[4] Data were analyzed using SAS Enterprse Guide version 7.1 (SAS Institute A/S, Carey, NC) and we used SigmaPlot version 12.5 (Systat Software, San Jose, CA) for plotting the ROC curve.2.7. Ethics and consentThe study was approved by the Danish Health and Medicines Authority (record no. 3-3013-135/1/) and by the Danish Data Protection Agency (record no. 01706 hvh-2012-016). Danish legislation does not require informed consent for register-based studies or ethical approval.3. ResultsA total of 8878 CSF analyses were reviewed. Of these, 7819 were excluded; 6248 due to fewer than 10 × 106 leukocytes/L, 697 due to age <16 years: 451 due to resampling within 90 days, 205 due to lack of leukocyte count analysis, 94 of samples was related to a neurosurgical procedure, 67 sampled were duplicates; and 28 with unavailable patient records: 22 due to blood contamination and 8 due to mislabeling.A total of 1058 CSF samples from 1054 patients were included in the analysis. The median age was 50 (interquartile range: 36–67) and 53% were male. Eighty-one different diagnoses were related to an elevated CSF leukocyte count, apart from unknown causes (Table (Table1).1). Viral meningitis was the most prevalent cause (18.6%), followed by BM (16.7%), Lyme neuroborreliosis (8.5%), demyelinating disease (6.3%), and stroke (4.9%).Table 1 Table 1Causes of cerebrospinal fluid pleocytosis and associated cell counts.3.1. Demographic data and supplemental CSF biochemistryData on patient age, sex, CSF leukocyte differential counts, protein levels, and glucose levels are presented in Table Table22.Table 2 Table 2Cerebrospinal fluid analysis characteristics according to disease categories.3.2. Disease categoriesThe distribution between the 6 disease categories is shown in Table Table3.3. Infections were the most common cause of CSF pleocytosis (61.3%) followed by miscellaneous causes (12.7%), vascular (9.7%), neurodegenerative (7%), neoplastic (5%), and inflammatory causes (4.2%). Only infections presented with CSF leukocyte counts >10,000 × 106/L and infections were also the most frequent cause of disease associated with a CSF leukocyte count >100 × 106/L, representing 82.6%. Among cases with CSF leukocyte counts <100 × 106/L infections constituted 43.3% of the diagnoses.Table 3 Table 3Distribution leukocyte count levels in cerebrospinal fluid according to disease categories.Eighty-one cases (7.7%) presented with CSF pleocytosis due to infections with a focus outside the CNS such as pneumonia, endocarditis, and septicemia. No microorganisms were detected and the samples assumed to represent sterile inflammation secondary to other infection.In 32 cases (3%), no diagnosis was established. These cases were labeled “unknown.”3.3. InfectionsInfectious causes of CSF pleocytosis were further divided into the following groups: acute bacterial, chronic bacterial, fungal, and viral CNS infections and inflammation secondary to an infectious focus outside the CNS (extra-CNS). Only 2 cases of parasitic CNS infections were identified and they were not included in this context. Data on CSF biochemistry, age, and sex for these groups are presented in Table Table44.Table 4 Table 4Cerebrospinal fluid analysis characteristics according to infectious categories.Since BM is arguably the most urgently treatable cause of CNS inflammation, we evaluated cut-offs for CSF leukocyte count to discriminate between BM and other CNS disease. We performed an ROC analysis estimating the probability of BM versus all other causes (Fig. (Fig.1)1) and versus other infectious causes only (Fig. (Fig.22).Figure 1 Figure 1Probability of bacterial meningitis versus all other diagnoses by receiver operating curve.Figure 2 Figure 2Probability of bacterial meningitis versus other infectious diagnoses by receiver operating curve.The first ROC curve (Fig. (Fig.1)1) yielded an area under the curve (AUC) of 0.92. Calculating the Youden J index suggested a leukocyte count of 414 as cut-off for diagnosing bacterial infection versus all other causes and it resulted in a sensitivity of 81%, a specificity of 89%, a positive predictive value off 62%, and a negative predictive value off 96% (Table (Table55).Table 5 Table 5Probability of bacterial meningitis.The second ROC curve (Fig. (Fig.2)2) yielded an AUC of 0.90. Calculating the Youden J index suggested a leukocyte count of 421 as cut-off for diagnosing bacterial infection versus other infectious causes and it resulted in a sensitivity of 80%, a specificity of 86%, a positive predictive value off 71%, and a negative predictive value off 92% (Table (Table55).We further investigated 3 different cut-offs for CSF leukocyte count: ≥100, ≥500, and ≥1000, to discriminate BM from all other causes and from other infectious causes, see Table Table5.5. We found that sensitivity decreased and specificity increased with increasing CSF leukocyte count, as expected.4. DiscussionThe presented data illustrate the variety of diseases that may be associated with an inflammatory process within the CNS. To our knowledge, no recent studies have presented data related to the causes of CNS inflammation on such a large number of cases.Infections presented with the highest CSF leukocyte counts and were also the most frequent cause of CSF pleocytosis, representing 61.4% of cases. Among patients presenting with CSF leukocyte counts below 100 × 106/L, however, the causes were more heterogeneous and noninfectious causes represented 56.7% of cases.Our finding of a considerable number of patients with sterile CSF inflammation secondary to systemic infections such as septicemia or pneumonia is not surprising since these infections may precipitate BM. Thus, these cases may represent a continuum toward the group of patients with culture negative meningitis although not treated as such. Among children, it is a well-described phenomenon that urinary tract infections may also present with sterile CSF pleocytosis.[5] Also a number of patients did not receive a diagnosis related to the presence of CSF inflammation. This group labeled “unknown” may represent a group of patient without CNS inflammatory disease albeit with CSF biochemistry differing from the normal population—thus normal variation.Based on the CSF leukocyte count alone, which is often the most quickly obtainable result, we calculated that CSF leukocyte counts below 414 × 106/L had a high specificity (89%) and negative predictive value (96%) for BM in this population. These results were largely unaffected when performed against all included diagnoses compared to the selection of infectious conditions only. Determining the “optimal cut-off” for diagnosis BM requires weighting the pros and cons. BM is a serious disease with mortality up to 30%[6] and failing to make a rapid and correct diagnosis might have fatal consequences. The cut-off suggested by Youdens index results in an NPV of 96%. Most clinicians might find this inadequate, as 4% would be left undiagnosed. Lowering the cut-off to 100 would improve the NPV with 2%, but reduce the PPV with nearly 30%. The reduced PPV could lead to inappropriate antibiotic use, which in this case would be of little concern due to the limited number of cases. Although the high AUC of ≥0.90, the cell count cannot alone confirm or discharge the diagnosis of BM. As shown by Hasbun et al,[7] predictive scores defined to safely identify urgently treatable CNS disease need also to include the clinical situation, the presentation of the patient and further biochemical results.Our cut-offs determined by Youden (414 and 421 × 106 leukocytes/L) are in agreement with previous studies in children, albeit our cut-offs were significantly lower, and we reached a similar specificity without including any other patient data.[8,9] The lower CSF leukocyte threshold is likely due to our adult population.A number of limitations should be acknowledged due to the retrospective collection of data and diagnoses obtained from discharge records. We did, however, attempt to minimize these weaknesses by reviewing the records twice and drawing other clinical and diagnostic data into consideration where available. A recent Danish study including also our population specifically evaluated the validity of the diagnosis herpes simplex encephalitis in the Danish National Patient Registry and found that this diagnosis could only be confirmed in ∼51% of cases based on strict inclusion criteria and another ∼8% considered to be probable cases, meaning more than 40% were incorrectly diagnosed.[10] The diagnostic confusion primarily involves the diagnosis of viral meningitis and the continuum toward viral encephalitis, but also culture negative BM may be difficult to define.[6]Many of the patients presenting with an elevated leukocyte count would most likely have received initial treatment for a suspected infection prior to receiving the final diagnosis. Thus, the number of infectious conditions may be underestimated. Also, it is well known that a few cases of severe CNS infections, that is, BM may present without CSF inflammation but often with positive microscopy. These patients presenting with <10 (×106/L) CSF leukocytes are not included in the presented data. Even though likely to be few, we cannot for certain exclude that they would affect our data.New diagnostics and improved methodologies could increase the number of correct diagnoses. Interestingly, we did not find any cases receiving a final diagnosis of autoimmune encephalitis, although 2 cases were suspected as having a postinfectious autoimmune condition and paramalignant limbic encephalitis, respectively. These diagnoses were not confirmed. Although autoimmune brain disease does not necessarily present with pleocytosis, the lack of cases in our data set could imply that this disease has received focus only in the most recent years and our data may include misdiagnosed cases.Even though this study is not a guideline to diagnostics of CNS diseases, we believe that our data may serve as a reminder to clinicians of what diseases and disease categories to suspect when patients present with CSF biochemistry indicating CNS inflammation.AcknowledgmentThe authors wish to thank Annette Farre for assisting in retrieving CSF analyses from the biochemistry databases LABKA I and LABKA II.FootnotesAbbreviations: AUC = area under the curve, BM = bacterial meningitis, CNS = central nervous system, CSF = cerebrospinal fluid, ROC = receiver operating characteristic.Joined first authorship between Gertrud Baunbæk Egelund and Gideon Ertner.CTB has received funding from the OTICON foundation for the studies of “cochlear damage in central nervous system infections.” This funding is not related to the present. No conflicts of interest and no other disclosures.TLB is the Assistant Editor of the Danish Medical Journal and reports personal fees from Bristol Myers Squibb, personal fees from Gilead, personal fees from Bristol Myers Squibb, personal fees from Bristol Myers Squibb, personal fees from GSK, nonfinancial support from Bristol Myers Squibb, nonfinancial support from Gilead, personal fees from Abbvie, outside the submitted work. The remaining authors have no conflicts of interest to disclose.Article informationMedicine (Baltimore). 2017 May; 96(18): e6686.Published online 2017 May 5. doi: 10.1097/MD.0000000000006686PMCID: PMC5419909PMID: 28471963Gertrud Baunbæk Egelund, MD,a,∗ Gideon Ertner, MD,a Kristina Langholz Kristensen, MD,a Andreas Vestergaard Jensen, MD,a Thomas L. Benfield, MD, DMSc,b and Christian T. Brandt, MD, DMScaMonitoring Editor: Jesper Kers.aDepartment of Pulmonary and Infectious Diseases, Nordsjællands Hospital, University of Copenhagen, HillerødbDepartment of Infectious Diseases, Hvidovre Hospital, University of Copenhagen, Hvidovre, Denmark.∗Correspondence: Gertrud Baunbæk Egelund, Nordsjællands Hospital, University of Copenhagen, Hillerød, Dyrehavevej 29, 3400 Hillerød, Denmark (e-mail: moc.liamg@nesdunkabdurtreg).Received 2016 Nov 28; Revised 2017 Mar 24; Accepted 2017 Mar 28.Copyright © 2017 the Author(s). Published by Wolters Kluwer Health, Inc.This is an open access article distributed under the Creative Commons Attribution-NoDerivatives License 4.0, which allows for redistribution, commercial and non-commercial, as long as it is passed along unchanged and in whole, with credit to the author. http://creativecommons.org/licenses/by-nd/4.0Articles from Medicine are provided here courtesy of Wolters Kluwer HealthReferences[1] Boyd W. Physiology and Pathology of the Cerebrospinal Fluid. New York, NY: The Macmillan Company; 1920.[2] Lundie A, Thomas DJ, Fleming S. Cerebro-spinal meningitis: diagnosis and prophylaxis is lumbar puncture justifiable? Br Med J 1915;1:628–9. [PMC free article] [PubMed][3] Fishman R. Cerebrospinal Fluid in Diseases of the Nervous System. 2nd ed.Philadelphia, PA: WB Saunders; 1992.[4] Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32–5. [PubMed][5] Syrogiannopoulos GA, Grivea IN, Anastassiou ED, et al. Sterile cerebrospinal fluid pleocytosis in young infants with urinary tract infection. Pediatr Infect Dis J 2001;20:927–30. [PubMed][6] Baunbaek-Knudsen G, Solling M, Farre A, et al. Improved outcome of bacterial meningitis associated with use of corticosteroid treatment. Infect Dis (Lond) 2016;48:281–6. [PubMed][7] Hasbun R, Bijlsma M, Brouwer MC, et al. Risk score for identifying adults with CSF pleocytosis and negative CSF Gram stain at low risk for an urgent treatable cause. J Infect 2013;67:102–10. [PMC free article] [PubMed][8] Nigrovic LE, Kuppermann N, Malley R. Development and validation of a multivariable predictive model to distinguish bacterial from aseptic meningitis in children in the post-Haemophilus influenzae era. Pediatrics 2002;110:712–9. [PubMed][9] Bonsu BK, Ortega HW, Marcon MJ, et al. A decision rule for predicting bacterial meningitis in children with cerebrospinal fluid pleocytosis when gram stain is negative or unavailable. Acad Emerg Med 2008;15:437–44. [PubMed][10] Jorgensen LK, Dalgaard LS, Ostergaard LJ, et al. Validity of the coding for herpes simplex encephalitis in the Danish National Patient Registry. Clin Epidemiol 2016;8:133–40. [PMC free article] [PubMed] |
|
|
|
Hypertrglyridemia causes |
This site is intended for healthcare professionalsMedscape LogoDrugs & Diseases > EndocrinologyHypertriglyceridemiaUpdated: Jun 20, 2018 Author: Mary Ellen T Sweeney, MD; Chief Editor: Romesh Khardori, MD, PhD, FACP more...Share FeedbackSECTIONSPractice EssentialsHypertriglyceridemia, a condition in which triglyceride levels are elevated, is a common disorder in the United States. It is often caused or exacerbated by uncontrolled diabetes mellitus, obesity, and sedentary habits, all of which are more prevalent in industrialized societies than in developing nations. In epidemiologic and interventional studies, hypertriglyceridemia is a risk factor for coronary artery disease (CAD).Hyperlipoproteinemia is a metabolic disorder characterized by abnormally elevated concentrations of specific lipoprotein particles in the plasma.Hyperlipidemia (ie, elevated plasma cholesterol or triglyceride levels or both) is present in all hyperlipoproteinemias. The primary form includes chylomicronemia, hypercholesterolemia, dysbetalipoproteinemia, hypertriglyceridemia, mixed hyperlipoproteinemia, and combined hyperlipoproteinemia. Other diseases, such as diabetes mellitus, pancreatitis, renal disease, and hypothyroidism, cause the secondary form.Signs and symptomsHypertriglyceridemia is usually asymptomatic until triglycerides are greater than 1000-2000 mg/dL. Signs and symptoms may include the following:GI: Pain in the mid-epigastric, chest, or back regions; nausea, vomitingRespiratory: DyspneaDermatologic: XanthomasOphthalmologic: Corneal arcus, xanthelasmasSee Clinical Presentation for more detail.DiagnosisOn examination, findings may be normal, or they may include the following:GI: Tenderness to palpation over mid-epigastric or upper right/left quadrants; hepatomegalyDermatologic: Eruptive xanthomas on back (see the image below), buttocks, chest, proximal extremities; palmar xanthomas in dysbetalipoproteinemiaEruptive xanthomas on the back of a patient admittEruptive xanthomas on the back of a patient admitted with a triglyceride level of 4600 mg/dL and acute pancreatitis.View Media GalleryCardiovascular: Decreased pedal pulses or ankle/brachial index in presence of peripheral vascular diseaseOphthalmologic: Corneal arcus, lipemia retinalis, xanthelasmas [1]Neurologic: Memory loss, dementia, and depression in the presence of chylomicronemia syndromeTestingLaboratory studies used to evaluate hypertriglyceridemia include the following:Lipid analysisChylomicron determinationFasting blood glucose levelTSH levelUrinalysisLiver function studiesProceduresIf the diagnosis of eruptive xanthomas is in doubt, obtaining a biopsy of the suspicious lesions will reveal accumulations of fat (not cholesterol). A biopsy of cutaneous lesions suspected to be either planar or tuberous xanthomas will reveal cholesterol deposition.See Workup for more detail.Fredrickson classificationHyperlipidemia has been defined by the Fredrickson classification, which is based on beta-quantification, a process involving ultracentrifugation followed by electrophoresis. [2] In this system, shown in Table 1, below, all categories except type IIa are forms of hypertriglyceridemia.Table 1. Fredrickson Classification of Hyperlipidemia (Open Table in a new window)TypeSerum ElevationLipoprotein ElevationICholesterol and triglyceridesChylomicronsIIaCholesterolLDLIIbCholesterol and triglyceridesLDL, VLDLIIICholesterol and triglyceridesIDLIVTriglyceridesVLDLVCholesterol and triglyceridesVLDL, chylomicronsIDL = intermediate-density lipoprotein; LDL = low-density lipoprotein; VLDL = very low-density lipoprotein.Source: Fredrickson DS, Lees RS. A system for phenotyping hyperlipidaemia. Circulation. Mar 1965;31:321-7. [2]Type I is a rare disorder characterized by severe elevations in chylomicrons and extremely elevated triglycerides, always reaching well above 1000 mg/dL and not infrequently rising as high as 10,000 mg/dL or more. It is caused by mutations of either the lipoprotein lipase gene (LPL), which is critical for the metabolism of chylomicrons and very low-density lipoprotein (VLDL), or the gene's cofactor, apolipoprotein (apo) C-II.Counterintuitively, despite exceedingly high elevations of triglyceride and, in some cases, of total cholesterol, these mutations do not appear to confer an increased risk of atherosclerotic disease. This fact may have contributed to the unfounded belief that hypertriglyceridemia is not a risk factor for atherosclerotic disease. Although chylomicrons contain far less cholesterol than other triglyceride-rich lipoproteins do, when serum triglyceride levels are severely elevated, cholesterol levels can also be quite high.Type IIb is the classic mixed hyperlipidemia (high cholesterol and triglyceride levels), caused by elevations in low-density lipoprotein (LDL) and VLDL.Type III is known as dysbetalipoproteinemia, remnant removal disease, or broad-beta disease due to an individual’s decreased ability to convert VLDL and intermediate-density lipoprotein (IDL), a VLDL remnant, to LDL particles in the blood and because of a decreased clearance of chylomicron remnants. [3] Typically, patients with this rare condition have elevated total cholesterol (range, 300 600 mg/dL) and triglyceride levels (usually >400 mg/dL; may exceed 1000 mg/dL), [2] and these individuals are easily confused with patients with type IIb hyperlipidemia. Patients with type III hyperlipidemia have elevations in IDL.Dysbetalipoproteinemia is the result of 2 "hits." [3] First, most affected individuals are homozygous for the apo E isoform, apo E2. Second, affected individuals usually have a metabolic disorder such as diabetes, obesity, or hypothyroidism. Consequently, those who are homozygous for apo E2 have a 1-2% risk of developing dysbetalipoproteinemia, and they are at high risk for atherosclerotic cardiovascular and peripheral vascular disease. [3] The condition responds well to treatment of the causative medical condition and to lipid-lowering medications.Type IV is characterized by abnormal elevations of VLDL, and triglyceride levels are almost always less than 1000 mg/dL. Serum cholesterol levels are normal.Type V is characterized by elevations of chylomicrons and VLDL. Triglyceride levels are invariably greater than 1000 mg/dL, and total cholesterol levels are always elevated. The LDL cholesterol level is usually low. Given the rarity of type I disease, when triglyceride levels above 1000 mg/dL are noted, the most likely cause is type V hyperlipidemia.Triglyceride levels greater than 1000 mg/dL increase the risk of acute pancreatitis, and because triglycerides are so labile, levels of 500 mg/dL or greater must be the primary focus of therapy. If a patient also has a high risk for a cardiovascular event, LDL-lowering therapy should be considered.ManagementNonpharmacotherapyNonpharmacologic management of hypertriglyceridemia is generally the initial treatment for patients with this condition. This primarily involves lifestyle modifications such as diet, exercise, weight reduction, smoking cessation, and limiting alcohol intake.PharmacotherapyMedications used in the management of hypertriglyceridemia include the following:Fibric acid derivatives (eg, gemfibrozil, fenofibrate)Niacin (slow-release, immediate-release, extended-release formulations)Omega-3 fatty acids (eg, omega-3-acid ethyl esters)HMG-CoA reductase inhibitors (eg, atorvastatin, fluvastatin, pitavastatin, pravastatin, lovastatin, simvastatin, rosuvastatin)Surgical optionIn general, surgical intervention is not necessary to treat hypertriglyceridemia. Plasmapheresis can be used in the setting of severe hypertriglyceridemia to reduce triglycerides in the acute setting. Ileal bypass surgery has been shown to improve all lipid parameters but should be reserved for severe hypertriglyceridemia refractory to all treatment.See Treatment and Medication for more detail.PathophysiologyTriglyceridesTriglycerides are fats consisting of 3 fatty acids covalently bonded to a glycerol molecule. These fats are synthesized by the liver or, in the case of those derived from dietary sources, are ingested by the liver (as described below); the triglycerides are subsequently transported throughout the circulation by triglyceride-rich lipoproteins.By dry weight, triglycerides make up approximately 86%, 55%, and 23% of chylomicrons, very low-density lipoproteins (VLDLs), and intermediate-density lipoproteins (IDLs), respectively, [4] as represented in the image below. Triglycerides are present in low-density lipoprotein (LDL) and high-density lipoprotein (HDL), but in much smaller quantities of 10% or less.The following image shows composition of triglyceride-rich proteins.Composition of triglyceride (TG)-rich lipoproteinsComposition of triglyceride (TG)-rich lipoproteins. IDL = intermediate-density lipoprotein; VLDL = very low-density lipoprotein.View Media GalleryTriglyceride-rich lipoproteins come from 2 sources, often described as the endogenous and exogenous pathways. In the exogenous pathway, dietary fats (triglycerides) are hydrolyzed to free fatty acids (FFAs) and monoglycerides and are absorbed, with cholesterol, by intestinal cells. They are then reesterified and combined with apolipoproteins and phospholipids to form a nascent chylomicron, a process requiring microsomal triglyceride transfer protein (MTP). The initial apolipoproteins are apolipoprotein (apo) A, which are soluble and can transfer to HDL; and apo B48, a structural apolipoprotein that is not removed during catabolism of the chylomicron. Chylomicrons enter the plasma via the thoracic duct, where they acquire other soluble apolipoproteins, including apo CI, apo CII, apo CIII, and apo E, from HDL.VLDLs and apolipoproteinsVLDLs are produced by a process analogous to the exogenous pathway. Triglycerides may derive from de novo FFA synthesis in the liver and are metabolized by lipoprotein lipase to IDL, also called VLDL remnants. Lipoprotein lipase hydrolyzes triglycerides, releasing FFAs, which are taken up by myocytes and hepatocytes. Some apo Cs, phospholipids, and apo Es are lost, and triglycerides are transferred to HDL in exchange for cholesterol esters. IDL is, thus, cholesterol-enriched and triglyceride-poor compared to unmetabolized VLDL. As IDL is metabolized by hepatic lipase to LDL, the remaining surface apolipoproteins are lost. [5, 3, 6]Triglycerides may also derive from the uptake of remnant chylomicrons, VLDL, or FFAs from the plasma. Precursor VLDL combines triglycerides, the structural or transmembrane apo B100, and phospholipids, as well as cholesterol and some apo Cs and Es. The formation of the immature VLDL requires microsomal transfer protein (MTP). Once secreted into the plasma, VLDLs acquire more apo Cs and Es.Apo EsApoEs are ligands that have greater affinity for the LDL receptor than does apo B100. In fact, the LDL receptor is more accurately designated the B/E receptor. Apo E also binds with high affinity to the LDL receptor-related protein, which takes up chylomicron remnants, VLDL, and IDL. In addition, apo E binds to cell-surface heparan sulfate proteoglycans (HSPGs), which assists in the hepatic uptake of remnant lipoproteins. [3]The apo E gene has been cloned, sequenced, and mapped to chromosome 19. Genetically altered apo E–deficient mice develop severe dyslipidemia with accelerated atherosclerosis, whereas transgenic mice overexpressing apo E appear to be protected from atherosclerosis. [7, 8] Apo E has 3 isoforms that are present in slightly varying proportions, depending on race and geographic location. [5] Apo E3 is the most prevalent allele and for that reason was considered the “wild type” allele from which apo E2 and apo E4 were derived. Newer data, however, suggest that apoA4 was the earliest form of the protein. [9]Most animals, including primates, possess an apo E4 equivalent. [10] Compared with apo E3, apo E2 has less affinity for the receptor, and apo E4 has more. The alleles differ in 2 amino acid positions, 112 and 158. Apo E2 is most commonly caused by cysteine substituted for arginine at position 158 in apo E3. In apo E4, an arginine is substituted for cysteine at position 112 in apo E3. The substitutions are recessive in that dysbetalipoproteinemia requires the presence of 2 apo E-2 isoforms. [10] Other very rare genetic variants of apo E exist, and several of these have been shown to have defective binding to the LDL receptor and LDL receptor-like protein. These variants act in a dominant fashion in that only 1 copy of apo E is necessary for susceptibility to development of type III hyperlipidemia.white populations, approximately 1% of these individuals are homozygous for apo E2 (“first hit”); however, only 10% of those will develop the condition. A “second hit” is necessary, most commonly metabolic abnormalities that cause increases in VLDL. [3] Other, less common genetic conditions can also predispose people to dysbetalipoproteinemia.More than 90% of patients with dysbetalipoproteinemia are homozygous for apo E2; the remainder have a rare, usually dominant, defect in apo E2. In addition to the apoE2 homology or defect, and combined with a metabolic condition, other genetic factors have been suggested that increase the likelihood of developing dysbetalipoproteinemia. Polymorphisms in the apo A5, lipoprotein lipase and apo C3 have all been mentioned as possible cofactors for the condition. [5]Accumulation of IDL is caused by the poor affinity of apo E2 to LDL receptors, whereas LDL uptake via apo B100 is unaffected. In fact, total cholesterol, LDL cholesterol, and apo B are usually low compared with those with apo E3. HDL cholesterol levels may be normal or decreased. The following 3 mechanisms have been postulated for the hypocholesterolemic effect of apo E2 [11] :Increased upregulation of LDL receptors due to decreased binding of lipoproteins containing apo E2Increased hepatic LDL uptake due to lower LDL receptor affinity of apo E2 and consequent decreased competition with the apo B100 born by LDL (its sole apolipoprotein)Apo E2 interference with lipolysis of VLDL to LDL [3]Chylomicron and VLDL metabolismAny disturbance that causes increased synthesis of chylomicrons and/or VLDLs or decreased metabolic breakdown causes elevations in triglyceride levels. That disturbance may be as common as dietary indiscretion or as unusual as a genetic mutation of an enzyme in the lipid metabolism pathway. Essentially, hypertriglyceridemia occurs through 1 of the following 3 processes [12] :Abnormalities in hepatic VLDL production, and intestinal chylomicron synthesisDysfunctional lipoprotein lipase -mediated lipolysisImpaired remnant clearanceAs shown in the images below, chylomicrons and VLDLs are initially metabolized by lipoprotein lipase, which hydrolyzes the triglycerides, releasing FFAs; these FFAs are stored in fat and muscle. With normal lipoprotein lipase activity, the half-lives of chylomicrons and VLDLs are about 10 minutes and 9 hours, respectively. Because of the large size of unmetabolized chylomicrons, they are unlikely to be taken up by macrophages, which are the precursors of foam cells. Foam cells promote fatty streak formation, the precursor of atherosclerotic plaque. Lipoprotein lipase activity produces chylomicron remnants that are small enough to take part in the atherosclerotic process. Chylomicron remnants are taken up by the LDL receptor or the LDL receptor-related protein. [13]Lipoprotein lipase (LPL) releases free fatty acidsLipoprotein lipase (LPL) releases free fatty acids (FFAs) from chylomicrons (chylo) and produces chylomicron remnants that are small enough to take part in the atherosclerotic process. Chol = cholesterol; TGs, TGS = triglycerides.View Media GalleryOnce very low-density lipoprotein (VLDL) has been Once very low-density lipoprotein (VLDL) has been metabolized by lipoprotein lipase, VLDL remnants in the form of intermediate-density lipoprotein (IDL) can be metabolized by hepatic lipase, producing low-density lipoprotein (LDL), or they can be taken up by the LDL receptor via either apolipoprotein B (apo B) or apo E. Chol = cholesterol; TGs = triglycerides.View Media GalleryVLDL remnants have 1 of 2 fates: they can be metabolized by hepatic lipase, which further depletes triglycerides, producing LDL, or they can be taken up by the LDL receptor via either apo B or apo E. VLDL remnants are not only triglyceride-poor, they are also cholesterol enriched, having acquired cholesterol ester from HDL via the action of cholesterol ester transfer protein (CETP), which facilitates the exchange of VLDL triglycerides for cholesterol in HDL. This pathway may promote HDL's reverse cholesterol transport activity, but only if VLDL and LDL return cholesterol to the liver. If these lipoproteins are taken up by macrophages, the CETP transfer results in increased atherogenesis.Chylomicron remnants, VLDL, VLDL remnants, and LDL are all atherogenic.EtiologyHypertriglyceridemia has many causes, including familial and genetic syndromes, metabolic disease, and drugs. Risks appear to include diet, stress, physical inactivity, and smoking.CHD risk factorsThe National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) identifies the 2 or more of the following as risk factors for coronary heart disease (CHD) that can lead to the need for more aggressive intervention [13] :Older age: In men, age 45 years or older, whereas in women, 55 years or older; the incidence of CHD is higher in the elderly than the young and in men more than women of the same ageFamily history of premature CHD: Paternal or male primary relative with myocardial infarction (MI) before age 55 years or sudden death, or in maternal or female primary relative before age 65 yearsActive tobacco useHypertension: Blood pressure higher than 140/90 mm Hg or current use of antihypertensive agentsLow level of high-density lipoprotein (HDL) cholesterol: Lower than 40 mg/dLGenetic causesAbnormalities of the enzyme pathway for chylomicron metabolism are the best-characterized genetic causes of hypertriglyceridemia. Lipoprotein lipase deficiency and apo C-II deficiency are caused by homozygous autosomal recessive genes present at conception.Type I hyperlipoproteinemia is the best-characterized genetic cause of hypertriglyceridemia and is caused by a deficiency or defect in either the enzyme lipoprotein lipase or its cofactor, apo C-II. Lipoprotein lipase hydrolyzes triglycerides in chylomicrons and very low-density lipoprotein (VLDL), releasing free fatty acids. The enzyme is found in the endothelial cells of capillaries and can be released into the plasma by heparin. Lipoprotein lipase is essential for the metabolism of chylomicrons and VLDL, transforming them into their respective remnants. Apo C-II, an apolipoprotein present in both chylomicrons and VLDL, acts as a cofactor in the action of lipoprotein lipase.The above pathway is affected by other genetic disorders, particularly type 1 or type 2 diabetes, because lipoprotein lipase requires insulin for full activity. That is, a secondary cause of hyperlipidemia, “second hit,” must be present for the dysbetalipoproteinemia to develop. In addition, the patient may be taking medications, such as protease inhibitors or tricyclic antidepressants, that exacerbate hyperlipidemia.Two more recently described syndromes include mutations in ApoAV leading to a truncated ApoAV devoid of a lipid-binding domain and glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GP1HBP1) causing decreased binding to LPL and reduced hydrolysis of chylomicrons. [14]Genetic predisposition for dysbetalipoproteinemia is present in approximately 1% of the population, but only 1-2% of individuals with apolipoprotein (apo) E-2 actually develop this condition. More than 90% of patients with dysbetalipoproteinemia are homozygous for apo E2. Extremely rare forms are associated with other genetic mutations in the apo E gene or the complete absence of apo E.Familial combined hyperlipidemia and familial hypertriglyceridemiaTwo triglyceride disorders, familial combined hyperlipidemia and familial hypertriglyceridemia, are genetically controlled, but the mechanisms are not clearly defined but are likely associated with overproduction and decreased of apo B–containing particles.Familial combined hyperlipidemia is an autosomal dominant disorder characterized by patients and their first-degree relatives who may have either isolated triglyceride or low-density lipoprotein (LDL) cholesterol elevations or both. Diagnosis of the disorder in a particular patient requires a family history of premature coronary artery disease (CAD) in 1 or more first-degree relatives and a family history for elevated triglycerides with or without elevated LDL cholesterol levels. The diagnosis is important for prognosis; 14% of patients with premature CAD have familial combined hyperlipidemia. [15]Familial hypertriglyceridemia is also an autosomal dominant trait. [16] These patients and their families have isolated triglyceride elevations and may have an increased risk of premature CAD.Genetic susceptibility factor effectsKnown genetic susceptibility factor effects account for approximately 10-15% of the trait variances in blood lipid levels (LDL cholesterol, HDL cholesterol, triglycerides). [17] Genome-wide association studies (GWAS) have identified several loci associated with blood lipid traits, including hypertriglyceridemia. [18]Hypertriglyceridemia is associated with several genes (in aggregate) including apoAV, GCKR, LPL, and APOB. [19] Patients with single nucleotide polymorphisms (SNPs) 40 kilobases (kb) from TRIB1 (a gene known to be strongly associated with dyslipidemia) have abnormal levels of triglycerides, as well as HDL cholesterol and LDL cholesterol. [20] In a large study of Japanese and Korean individuals, investigators reported that genetic variants of APOA5 (-1131T→C polymorphism [rs662799]) and BTN2A1 (C→T polymorphism [rs6929846]) synergistically affect the prevalence of dyslipidemia in East Asian populations and metabolic syndrome in Japanese individuals. [21]Certain genetic variants can further predispose a patient with hypertriglyceridemia and certain environmental factors to consequences, such as CAD and MI. For example, the genetic variant R952QP of LRP8 (a gene at 1p31-32 that is associated with familial and premature CAD as well as high-level platelet activation) is associated with high triglyceride levels in patients who are have a history of being overweight, smoke, and have premature CAD/MI. [22]Metabolic causesUncontrolled diabetes mellitus, both type 1 and type 2, is one of the most common causes of hypertriglyceridemia, and it is often severe in patients presenting with ketosis. Patients with type 1 diabetes mellitus are insulin deficient, and lipoprotein lipase is largely ineffective. Control of these patients' diabetes mellitus with insulin will restore lipoprotein lipase function, reducing triglyceride levels and restoring diabetes mellitus control.In patients with uncontrolled type 2 diabetes mellitus and hyperinsulinemia, triglycerides are elevated for several reasons, including the following:Lipoprotein lipase is less effective in the insulin-resistant stateOverproduction of VLDL by the liver is common in patients with diabetes who are often overweightDiabetes mellitus is one of the conditions that leads to incomplete metabolism of VLDL, causing increased remnant VLDL or IDL observed in dysbetalipoproteinemia (see Dysbetalipoproteinemia).Mild to moderate elevations in triglycerides are common in obese patients, largely secondary to reduced efficacy of LPL and overproduction of VLDL.Hypothyroidism commonly causes LDL cholesterol elevations, but it also may lead to mixed hyperlipidemia or isolated triglyceride elevations. Reduced hepatic lipase activity slows VLDL remnant catabolism. As with diabetes mellitus, untreated hypothyroidism may cause dysbetalipoproteinemia in patients with homozygous apo E-2.Nephrotic syndrome is thought to increase hepatic synthesis of VLDL and may also slow catabolism of both LDL and VLDL. As in hypothyroidism, elevated LDL cholesterol levels are more common in this condition, but mixed hyperlipidemia or isolated triglyceride elevations may be observed. Higher levels of proteinuria are correlated with more severe hyperlipidemia.DrugsMedications that can cause hypertriglyceridemia include the following:High-dose thiazide diuretics or chlorthalidoneHigh-dose beta-adrenergic blocking agents, excluding those with intrinsic sympathomimetic activityUnopposed oral estrogen replacement therapyOral contraceptives with high estrogen contentTamoxifenGlucocorticoidsOral isotretinoinAntiretroviral therapy (including some protease inhibitors, nonnucleoside reverse transcriptase inhibitors)Atypical antipsychoticsOther causesExcessive alcohol intake and high-carbohydrate diets (>60% of caloric intake) are frequent causes of hypertriglyceridemia.Acute pancreatitis may cause substantial elevations in triglycerides by unknown mechanisms. However, much more frequently, severe hypertriglyceridemia causes acute pancreatitis. In patients presenting with acute pancreatitis and triglycerides greater than 1000 mg/dL, it is prudent to not assume that the triglycerides are the cause of the pancreatitis. Other causes, such as common bile duct obstruction and alcoholism, must be considered as possible etiologies.A study by Zhang et al indicated that obesity and uric acid elevation have a strong additive interaction that increases the risk for nonalcoholic fatty liver disease and hypertriglyceridemia. The interaction was reportedly responsible for 27% and 26% of the expanded hypertriglyceridemia risk in men and women, respectively. [23]In pregnant patients with a history of mildly to moderately elevated triglycerides in the nonpregnant state, hypertriglyceridemia (sometimes severe) may occur. Such patients should be monitored closely, particularly in the third trimester. In fact, simply looking for laboratory notation of lipemic serum in routine blood tests during pregnancy will avoid unexpected complications resulting from unrecognized and untreated hypertriglyceridemia during pregnancy. |
|
|
|
Q Charecteristic of psychotic depression a. Delusions of poverty b. Flight. Of ideas c.delusions of nihilism d. visual hallucination e. |
Q |
|
|
|
what is used for treating alcohol withdrawal seizures |
Q diazepam |
|
|
|
Q |
. Right ventricular MI a. Tall R wave in anterior leads in posterior MI b. Treated with high dose statin with in 24 hours c. Treated with enoxaparin after thrombolysis |
|
|
|
Can cardiomyopathy be a complication of of AF |
Q |
|
|
|
What are the causes tinnitus |
Q hypertension Q BPPV |
|
|
|
Q Thyrotoxicosis a. Gynaecomastia b. Prolactinaemia |
Q |
|
|
|
Q Fasting hypoglycemia a. Liver failure b. Addison’s diseases c. Prolactinoma |
Q |
|
|
|
Q Pressure ulcers a. Antibiotics b. Sedative c. Immobility d.sulphanylureas |
Causes for pressure ulcers |
|
|
|
PUO causes |
Q Thyrotoxicosis |
|
|
|
Can mannitol be used to treat hyperkalemia |
Q |
|
|
|
What does hypertension give rise to |
Systolic or diastolic HF |
|
|
|
Does creatinine level rise in early stage |
Q |
|
|
|
Vitamin D increases a. Osteoblast b. c. Osteoclast |
What are the functions of vitamin D |
|
|
|
Q Iron absorption💡 a. Ferritin b. Transferrin c. Hepacidin e. Divalent metal transporter |
What |
|
|
|
True ☺ MEMANTINE |
Memantine is a NMDA receptor blocker used to treat behavioural symptoms in dementia |
|
|
|
True about ☺ GALANTAMINE |
It is AN ACETYLCHOLINE ESTERS INHIBITOR |
|
|
|
BISPHOSPHONATES ARE EFFECTIVE FOR ALL 3 |
Hip Vertebral Non vertebral ( alendronate/ residronate / zoledronic acid / denodranate ) |
|
|
|
The frequency of giving them |
Zoledronic acid |
|
|
|
Restless leg syndrome |
Ropinirole is the best treatment . Can be associated with iron deficiency anemia . Symptom augmentation can happen with treatment. Symptoms cannot be seen with activities. Other treatments Levodopa Dopamine agonists Gabapentine Pregablin Clonazepam Opioids |
|
|
|
Frailty |
Gait speed is a measure of frailty. Week hand grip is a risk factor. Treatment Male gonadotropins for males Growth hormone for both Women are at high risk as their muscle mass is poor Vitamin D treatment has no place |
|
|
|
Hey do you know |
Glomerular sclerosis is a normal finding with aging |
|
|
|
What causes constipation |
Hyper/ hypocalcemia |
|
|
|
Do you know 🔃🆑 |
With aging urine concentrating ability of the kidneys is reduced |
|
|
|
Action tremors |
Phenytoin is associated with action tremors |
|
|
|
Rest tremor |
Movement of the contralateral side increases the rest tremor |
|
|
|
Rest tremor |
Movement of the contralateral side increases the rest tremor |
|
|
|
What are the controlled release preparations used to treat motor fluctuations in Parkinson disease |
Q |
Modes of treatment in Parkinson disease Levodopa Dopamine agonists MAO B INHIBITORS |
|
|
Compared to dementia ,Delirium |
Shows memory impairment - T Shows motor signs frequently - T |
|
|
|
DISPROPORTIONATE MEDIAL TEMPORAL LOBE ATROPHY |
Is seen ALZHEIMER DEMENTIA |
|
|
|
Acetylcholine esterse inhibitors are more useful in early dementia |
Not in advanced state |
|
|
|
In elderly Low protein Low water Low muscle High lipid |
Lipophilic drug levels stays long Diazepam Free fraction of the protein bound form rise Digoxin free fraction Warfarin free fraction Frusemide free fraction
|
|
|
|
With aging they become less bradycardic to |
BETS BLOCKERS |
|
|
|
What can be used to manage end of life care patients breathlessness |
Opioids Benzodiazepines O2 |
|
|
|
Can thyrotoxicosis be a cause for puo |
Q |
|
|
|
Risk factors for uv prolapse |
PID? EARLY MENARCHAE? |
|
|
|
Q Most osteoporotic fractures occur when BMD is in the osteoporosis range |
Q |
|
|
|
Can hypokalemia occur with SAH |
Q |
|
|
|
Other sites for erythema nodosum |
Q hands and thighs |
|
|
|
Commonest cause of hyperthyroidism in elderly |
Q |
|