Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
100 Cards in this Set
- Front
- Back
|
what are the VERY first vessels to come off the aorta?
|
'dem coronaries - RIGHT, and LEFT --> LAD/circumflex
|
|
|
this may seem odd, but when do the coronaries fill will blood?
|
excellent question - during DIASTOLE because that's when the heart muscle relaxes and there's room to fill dem vessels up
|
|
|
do myocardial cells require oxygen for contraction or relaxation?
|
TRICK QUESTION: both!
- O2 extraction is always maxed out so the only way to increase O2 to tissues is to increase flow |
|
|
what do those epicardial arteries do in the heart?
|
- conduit vessels - NOT Responsible for resistance for flow when normal
|
|
|
what vessels in the heart create most of the resistance to flow in the heart?
|
- small, distal arterioles (DUH because arterioles have the highest resistance!!!)
- we can tolerate a shiz ton of blockage before sh@t hits the fan - arterioles try and dilate in order to make up for block |
|
|
when does myocardial ischemia occur? (obvious after a blockage, but in terms of when damage starts)
|
- when mVO2 > mO2 supply
(myocardial oxygen demand > myocardial oxygen supply) DECREASE SUPPLY OR INCREASE DEMAND - like in lo diastolic perfusion pressure, high coronary vascular resistance (b/c of compression or b/c of local metabolites, endothelial factors, or neural influence), or low O2 carrying capacity - increasing demand: increased wall tension, increased HR, increased contractility |
|
|
how is wall tension related to radius, wall thickness and o2 demand?
|
wall tension = pressure x radius / wall thickness
(so you might increase wall thickness in concentric hypertrophy to reduce wall tension like in aortic stenosis) |
|
|
what are the two main things that determine myocardial oxygen SUPPLY?
|
- coronary blood flow and O2 carrying capacity
|
|
|
what is the stupid formula for blood flow?
|
- flow = pressure/resistance
and in this case - pressure = coronary perfusion pressure resistance = coronary vascular resistance as you increase resistance, flow drops |
|
|
what is coronary perfusion pressure equal to? (think about vessel filling in the heart)
|
- LVEDP
|
|
|
when does coronary flow PEAK?
|
- right when diastole starts
|
|
|
if i were so say "autoregulatory resistance" what would I be talking about?
|
- metabolic control of vessel resistance controlled metabolically by O2, adenosine/ADP (MAJORRRR), NO, lactate, H+, histamine, bradykinin
|
|
|
how does the process of autoregulating vascular resistance work, exactly?
|
-heart is beating faster
-heart releases adenosine -adenosine triggers vascular endothelial cells to signal smooth muscle (if no normal endothelium, no response) -smooth muscle DILATES -more blood flow joy to the world |
|
|
in terms of regulating flow, O2 is a vaso_______
|
vasoCONSTRICTOR - makes sense - the more O2 you have, the less dilation you need, the less flow you need
as O2 levels drop in ischemia --> pre-capillary vasodilation and increased MI blood supply |
|
|
what is the most important autoregulatory mechanism?
|
- adenosine: SHORT 1/2 LIFE
- potent vasodilator: binds receptors on vascular smooth muscle and decreases Ca entry into cell - during hypoxemia: aerobic metab in mitochondria is inhibited --> accumulate ADP/AMP --> produce adenosine --> dilates arterioles --> increases coronary blood flow |
|
|
what happens in normal conditions when we increase perfusion pressure?
|
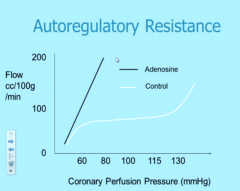
- normally, autoregulation prevents too much flow increase
- but we add a shiz ton of adenosine in this scenario - coronary flow skyrockets |
|
|
aside from awesome adenosine, what else autoregulates flow in coronaries?
|
- endothelial-derived factors
*dilators: Endothelial-derived relaxation factor = EDRF = nitric oxide (vasodilator, acts by ↑ cGMP), prostacyclin (increases cAMP) *constrictors: Endothelin-1 (leads to vasoconstriction during a heart attack → BAD!) |
|
|
what is coronary flow reserve?
|
- arteriolar autoregulatory vasodilatory capacity in response to increased MVO2 or pharm agents
- we can increase 4-5:1 experimentally, 2.25-3:1 clinically (max flow/baseline flow) |
|
|
what happens in the arterioles when an epicardial vessel stenoses?
|
- DILATION!
- as stenosis progresses, arteriolar dilation becomes chronic, decreasing potential to augment flow and thus decreasing CFR - endocardial CFF < epicardial CFR (think n comes before p) - as this maxes out, any further decrease in PP or increase in MVO2 --> ischemia (angina!) |
|
|
how does blood flow enter the ENDO vs EPICARDIUM?
|
- blood flows from muscle --> endocardium during diastole
- during systole - heart thickens & > flow to EPI and < to ENDO SYSTOLE loves EPI |
|
|
What is more vulnerable: epicardium or endocardium?
|
ENDOCARDIUM
because of that whole silly systole thing where EPI gets a sh@t ton of blood and ENDO doesn't - also: endo has greater shortening/thickening and higher wall tension --> increased MVO2 --> < collateral circulation --- decreased coronary flow reserve in endocardium |
|
|
does EPI or ENDOCARDIUM have a lower coronary flow reserve?
|
ENDOcardium because of increased mVO2 demand, less collaterals, systole where epi gets all the blood
|
|
|
im a healthy dude in my 30s. what risk factors would be concerning for me in terms of my risk for atherosclerosis?
|
- family history
- cigarette smoking - htn - hi lipids (LDL) - sedentary life - elevated homocysteine - inflammation - LP-a |
|
|
so i have an LDL of 200, i smoke and my dad had early CAD.... which of these is most concerning?
|
hi LDL
|
|
|
what is the relationship between amount of fat in the diet and LDL level?
|
linear! more fat we eat, more cholesterol we have
|
|
|
i'm an atherosclerotic plaque. how did i get to be me?
|
- fatty streak in a young adult
- then i became a soft atherosclerotic plaque with lots of fat (and i was very very vulnerable to fissuring/hemorrhage) - then all this complex stuff happened where i was interacting with substrate, circulating cells, platelets, macrophages, neurohumoral factors - all this stuff gave me a fibrous cap and smooth muscle started to migrate to the intima and produce a tough fibrous matrix which glued my cells together - while this transformation was happening, to compensate for all this fat all over the place, the outer wall of my vessel expanded in vessel remodeling |
|
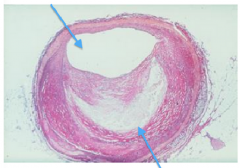
this shiz is bananas. what's the hap?
|
top arrow: lumen
bottom arrow: athlerosclerotic plaque crescent |
|
there's a lot of stuff going on here. what is that lots of stuff?
|

|
|
|
lady comes in with stable angina. what symptoms may she be complaining of?
|
- mid-substernal chest pain
- squeezing, pressure-like quality - builds peak and lasts 2-20 minutes - radiation to left arm, neck, jaw, back - associated with shortness of breath, sweating, nausea - exacerbated by exertion, cold, meals b/c <blood to heart because shunted to GI to digest - worse w/ stress - relieved by rest |
|
|
what test should i use to confirm stable angina?
|
stress test - patient w/ suspected angina is placed on a treadmill and asked to slowly increase walking pace; heart rate speeds up, ST segment depression
- thalium: nuclear tracer shows where blood is flowing during exercise vs rest |
|
|
now that we know that patient has angina, what do we do to treat this?
|
- RISK FACTOR MODIFICATION
- aspirin - inhibits platelet aggregation, decreases clot risk - decrease MVO2: NITRATES, beta blockers, Ca channel blockers, ACE inhibitors - angioplasty, stent etc or bypass |
|
|
how does atherosclerosis --> coronary thrombosis?
|
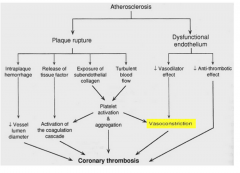
|
|
|
what three things do we worry about in the heart as a result of atherosclerosis after stable angina?
|
- unstable angina
- non-ST elevation MI - ST-elevation MI |
|
|
talk to me about unstable angina
|
- ST depression, T wave inversion or normal
- no enzyme release |
|
|
what are the characteristics of non-ST elevation MI?
|
non-STEMI
- ST depression, T wave inversion or NL - no Q waves - CPK, LDH + troponin release |
|
|
what are the characteristics of ST elevation MI?
|
- TRANSMURAL
- ST elevation - +Q waves - CPK, LDH + Troponin release |
|
|
what do you mean when you say someone has unstable angina?
|
- new onset angina
- increase in freq, duration, severity - decrease in exertion required to provoke - any prolonged episone 10-15minutes+ of chest discomfort - failure to abate w/ 2-3 nitroglycerins - onset at rest or awakening from sleep |
|
|
what are scary things that mean high risk angina?
|
- prolonged chest pain
- dynamic EKG changes ST depression w/ chest pain - age >65 - DM - LV systolic dysfunction - angina assoc w/ CHF, new murmur, arrhythmias or hypotension - elevated Troponin I or T |
|
|
what is the Wavefront phenomenon of ischemic evolution?
|
Endocardium to epicardium
If limited area of infarction → homeostasis achieved and patients do well If large area of infarction (>20% LV) → congestive heart failure If larger area of infarction (>40% LV) → hemodynamic collapse |
|
|
why do we care about troponin I levels?
|
- marker of MI
- HIGHLY SENSITIVE - tells us heart has been damaged - ONLY released w/ cell damage to heart muscle - may be elevated after prolonged subendocardial ischemia - causes of troponin elevation - even small amounts of ischemia, prolonged angina, prolonged tachy, CHF, hypoxia, aborted MI (lytic therapy) |
|
|
what are some bad things that happen after myocardial ischemia (symptoms and aftershocks)?
|
- chest pain
- systolic dysfunction --> decreased CO, decreased coronary perfusion pressure - diastolic dysfunction (loss of relaxation) - higher pressure for any volume, dyspnea, decreased pO2, decreased O2 delivery, increased wall tension --> increased MVO2 - all of these combined --> stimulation of sympathetic nervous system w/ subsequent catecholamine release: increased HR and blood pressure |
|
|
how to do treat an acute MI?
|
- ASA, thienopyridine, heparin, analgesia, O2
- reperfusion therapy - cath lab, throbolytic therapy - decreased MVO2 with nitrates, beta blockers, ACE inhibitors, and diuretics for high PCWP; for lo CO - pressors, IABP, early cath |
|
|
so i had an MI. how long do I have until irreversible damage occurs.
|
20 minutes! myocardial tissue injury begins (collaterals effect this)
|
|
|
how long after MI does chest pain set in?
|
late-ish, after permanent damage has been done
|
|
|
what are the earliest complications from MI?
|
- arrhythmias - heart block, brady, tachy (supraventricular, ventricular)
- hemodynamic disruption - CHF - hypotension, shock |
|
|
it's been 3-7 days since my MI. what is happening to my heart?
|
YELLOW SOFTENING
think - you have to watch these patients within the first week because they may suddenly die on you. - mechanical complications: papillary muscle rupture, free wall rupture, acute VSD, LV apical aneurysm - pericarditis - thromboembolism |
|
|
what is this "ischemic cascade" i have heard so much about?
|
- diastolic dysfunction - heart gets stiff, relaxation takes energy, pressure increases in LV during diastole
- localized systolic dysfunction - stops conducting properly - ischemic EKG changes - ST elevation - chest pressure - release of CPK/troponin enzymes |
|
|
i'm a heart cell and i just experience an ischemic event 9 weeks ago. let me tell you what i've been through
|
well,
- 20-30 minutes after i was irreversibly changed forever - within 24 hours i was all icky and gooey with coagulation necrosis - then from 5-7 days i was yellow and soft - after that i was remodeling myself until week 4 - then from weeks 6-8 i was fibrosing all over |
|
|
i am a clot. what is the place I should land to do the most damage?
|
- Left main coronary artery which supplies 2/3 of the myocardium
- LAD is pretty bad too - supplies 40% of LV (apex, septum, anterior wall), supplies 2/3 of septum, supplies most of conduction system below AV node --> LV failure, hi grade heart block, apical aneurysm formation, thrombo-embolic complications - RCA: < LV myocardium, but all RV, posterior 1/3 of septum, most of conduction system AT OR ABOVE AV node --> RV failure, brady, occasional mechanical problems |
|
|
what would you expect from an RCA MI?
|
- RV failure
- bradyarrhythmias because of involvement of SA and AV nodal arteries - mechanical stuff going on |
|
|
what would you expect from an LAD MI?
|
- LV failure
- high grade heart block b/c of lack of blood flow to His-Purkinje, no conduction from upper --> lower chambers - apical aneurysm form - thrombo-embolic complications |
|
|
leads I, II and AVF are looking a little funky with some ST elevation. what do you suspect is going on? what complications would you expect?
|
INFERIOR MI - RCA
- sinus brady - SA nodal artery and increased vagal tone - heart block b/c AV nodal artery occluded (1st degree AV block, Wenckebach 2nd degree A-V block, AV dissociation) - a fib due to LA stretch b/c LV has diastolic dysfunction - LVEDP increases --> increase LA pressure --> loss of atrial kick --> reduce CO - PVCs, ventricular tachy/fib via re-entry or increase in automaticity |
|
|
now there's something funky going on in the patient's EKG leads V1-4 where the ST segment is crazy high. what do you suspect is going on? what complications would you expect?
|
- ANTERIOR MI caused by LAD occlusion
- sinus tachy, reduced EF, reduced stroke volume --> need faster HR to compensate or pump tons of epi - heart block - his-purkinje: left or right bundle branch block, complete heart block - ventricular tachy/fib due to re-entry or increase in automaticity |
|
|
what are the hemodynamic consequences of MI?
|
- CHF - diastolic dysfunction, systolic dysfunction, increased LVEDP --> pulmonary congestion
- hypotension/shock: due to low preload, low stroke volume |
|
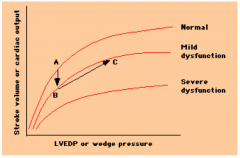
what's going on here?
|
CHF occurs when someone goes from A --> mild dysfunction curve B - patient needs to compensate by functioing at higher LVEDP/wedge pressure (C) in order to maintain the same CO
- give fluids - increase LVEDP and help maintain CO (B-->C) |
|
|
i'm a math kind of person. is there a way to determine how well someone's heart is doing based on the F-S curve that is a little more exact?
|
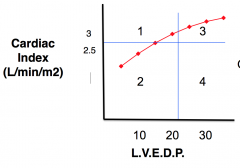
YES! Plot the F-S curve and then divide into quadrants 1, 2, 3, 4. You want to be in quadrant 1.
|
|
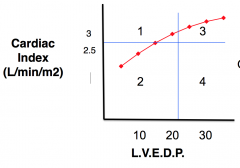
say i've plotted this F-S. what does each of those quadrants mean?
|
1: best prognosis - normal; low LVEDP, normal CI, not in CHF
2/3: intermediate prognosis (85%) survival; 2: could be someone w/ lo contractility, lo BP, lo CI, but not CHF because LVEDP not elevated; if you give this person fluid --> Q1 b/c no pulm edema since not in CHF 3: if you give Q2 too much fluid could end up here w/ high LVEDP --> transudation of fluids 4: cardiogenic shock (20-40% survive): low CO, high LVEDP --> lo BP w/ fluid in lungs --> can't give any more fluids |
|
|
what do you do for a person in cardiogenic shock who just had an MI?
|
- get artery open!
- supportive measures: inotropes, intra-aortic balloon pump, LV assistance devices - look for correctable causes - RV infarct, mechanical complications during days 3-7 |
|
|
what are some terrible mechanical complications that can occur after MI?
|
- rupture of free wall --> tamponade or pseudoaneurysm
- rupture of papillary muscle --> acute mitral regurg - rupture of septum --> acute VSD |
|
|
holy moly!!! the free wall ruptured... what's going to happen???
|
- cardiac tamponade: blood rushes into the pericardial sac; equalization of diastolic pressures, hypotension --> compression of heart, altered JVP --> JVD, clear lung fields, pulsus paradoxus
- pseudoaneurysm: localized, blood exiting LV is walled off and contained by pericardium, can go undetected --> enlarged cardiac silhouette, echo diagnosis, in pseudo broke thru all layers of heart and created a hole and pericardium is what keeps the blood |
|
|
JEEZ! my papillary muscles ruptured. what's going to happen now??
|
- acute mitral regurg --> systolic murmur, giant systolic V waves --> CHF --> decreased BP
(posterior leaflets most vulnerable) |
|
|
what does it mean when you say someone "developed giant v waves"
|
- when you measure wedge pressure, you would normally have a baby teeny v wave
- MR - GIANT v-wave think: MR. V is a big deal |
|
|
great, so now i have acute mitral regurg. how do you treat this?
|
- rapid diagnosis
- rx with AFTERLOAD reduction - inotropic support - intra-aortic balloon pump - surgical valve replacement |
|
|
OMG! i ruptured my intraventricular septum. what's going to happen to me?
|
- now i have an acute VSD --> L-R shunt
- abrupt onset of harsh systolic murmur + thrill - detected by O2 step up - normally RA and RV would have the same O2... not true here |
|
|
what do you do for someone with intraventricular septal rupture?
|
SAME AS MR
- rapidly diagnose - afterload reduction - favor forward flow w/ arterial vasodilator - inotropic support - contractility w/ dopa drug - intra-aortic baloon pump - surgical repair of ruptured septum |
|
|
what is this intra-aortic balloon pump you speak of?
|
- increases coronary flow during diastole
- decreases afterload during systole be deflating at onset - decreases myocardial ischemia - pump into femoral artery --> descending aorta; inflates and deflates depending on cycle - inflates/increases pressure in aorta during diastole --> more coronary blood flow and during systole it deflates --> vacuum that pulls blood from LV into aorta by reducing afterload |
|
|
apical aneurysm: who what when where why how
|
- LAD occlusion --> outpouching of LV from scar tissue at apex; w/o reperfusion --> apical aneurysm
- associated w/ large transmural antero-apical MI - BAD: makes LV bigger --> increased wall tension --> increased diastolic pressure - can --> apical thrombus from LV - ventricular arrhythmias - dyskinesis of apex - give anticoagulants |
|
|
right heart failure - causes and consequences
|
- RV infarct in RCA --> JVD w/ clear lungs, equal RA and PCW, ST elevation in R precordial leads
- give fluids |
|
|
what are the vascular changes associated with ischemic heart disease?
|
- fixed narrowing
- acute plaque changes - thrombosis - vasospasm |
|
|
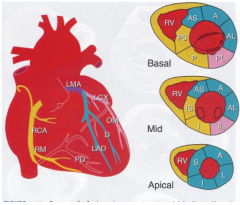
|
|
|
What is the structure of an atheromatous plaque?
|
Fibrous cap: outermost layer of plaque, contains smooth muscle cells, macrophages, foam cells, lymphocytes
Necrotic core: inner layer w/ cell debris, cholesterol crystals, foam cells, calcified structures |
|
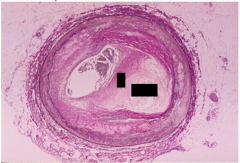
What is this?
|
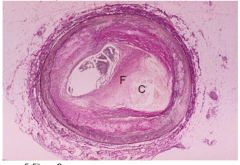
- histo slide of plaqueF: fibrous cap, C: necrotic lipid core
|
|
|
What makes a plaque likely to rupture?
|
- fibrous cap is thin
- necrotic core is filled with lipids and makes up larger percent (>40%) of plaque |
|
|
How much occlusion do you need for 1) significant lesion 2) critical lesion 3) stable angina.
|
1) significant: 50% reduction in diameter (=75% area reduction)
2) critical: 75% reduction in diam (90% area) 3) angina: 50-75% in diameter (75-90% area) |
|
|
What happens in unstable angina?
|
Plaque erosion
|
|
|
What happens during acute plaque changes?
|
- platelets aggregate and either: 1) plaque disruption --> healing --> fibrosis and lumen of vessel shrinks
2) acute plaque changes ----- erosion of endotehlium (erosion --> healing --> erosion --> healing) AND/OR plaque rupture --> thrombosis AND/OR hemorrhage into necrotic core AND/OR thrombosis of blood in lumen |
|
|
what is the difference between plaque rupture and plaque erosion?
|
- rupturing thru fibrous cap exposes blood to highly thrombotic core
- b/c of this, significant thrombosis >>> in RUPTURE than erosion |
|
|
what do plaques rupture?
|
- macrophages release metalloproteinases that --> destablization of plaque + digest collagen in fibrous cap
- vulnerable plaque more likely to rupture if: 1) - >40% of atherosclerotic lesion composed of core 2) thin fibrous cap 3) many macrophages 4) low smooth muscle in fibrous cap |
|
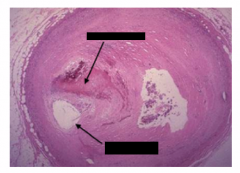
what happened here?
|
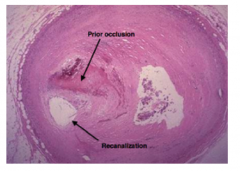
recanalization of vessel on its own following occlusion
|
|
|
what are the differences between transmural and subendocardial infarcts?
|
TRANSMURAL: ENTIRE WALL, due to plaque rupture and coronary thrombosis; Q waves on EKG; starts in sub-endocardial region (inner --> outwards)
SUBENDOCARDIAL: only inner 1/3 - 1/2 wall damaged, not limited to coronary branch zone; non-Q wave; related to global reduction in coronary blood flow |
|
|
what are the biochemical changes that occur mediately after an MI?
|
- rapid fall in ATP
- quick rise in lactate |
|
|
what is the time course following MI?
|
few minutes: cell swelling, mitochondrial swelling, glycogen depletion
20-40 minutes: loss of reversibility 6 hours: myocytes die in wavefront from subendocardium to subepicardium |
|
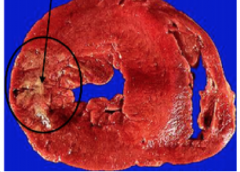
how long after MI was this?
|
4 days
|
|
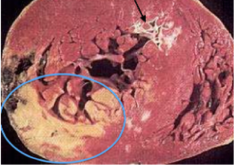
how long after MI was this?
|
several days
(note a prior infarct at the arrow) |
|
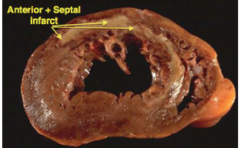
how long after MI was this?
|
several days
|
|
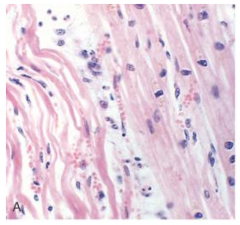
what is this? how long after MI does it occur?
|
wavy fibers - 1-3 hours
|
|
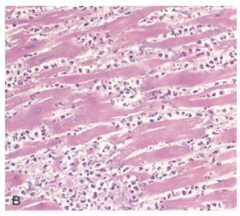
what is this? how long after MI does it occur?
|
loss of nuclei, loss of striations, lots of PMNs - 48 hours;
(4-12 hours: coagulative necrosis w/ hypereosinophilia, loss of striations) (24-48 hours: nuclear disappearance) |
|
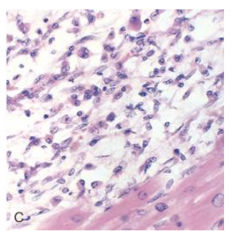
what is this? how long after MI does it occur?
|
4-6 days: macrophages
(3 days: proliferation of BVs) (3-7 days: highest risk for myocardial rupture w/ lots of soft tissue from necrosis and no scar tissue) |
|
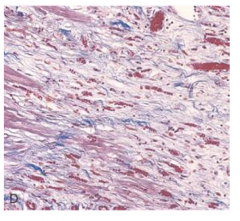
what is this? how long after MI does it occur?
|
granulation tissue forming - 1-4weeks
red areas are new capillaries; collagen and granulation tissue is indicative of healing |
|
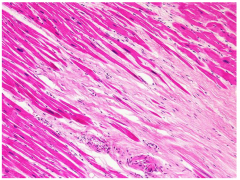
what is this? how long after MI does it occur?
|
6 weeks: advanced scarring; once scar is well healed you can't tell how old it is; this is a healed myocardiocyte w/ dark purple areas of replacement fibrosis and light areas of compensatory hypertrophy
|
|
|
what is myocytolysis?
|
vacuolar degeneration of mycardial cell, a degenerative change (often reversible) that happens in neighboring cells during infarct - found on the perimeter of MIs and in subendocardium
appearance: looks like nucleus is in an empty sarcolemmal tube; seen best in LV subendocardium and perimeter of MIs - these cells are still viable |
|
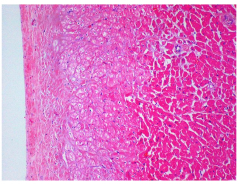
what happened to these cells?
|
lighter cells on the left got some O2 are are undergoing myocytolysis
|
|
|
describe the etiology of reperfusion injury.
|
- lethal injury to cells that were otherwise ok
- due to generation of reactive O2 species, intracell Ca+ overload, inflammation - contraction band necrosis occurs |
|
|
what is contraction band necrosis?
|
- manifestation of reperfusion in jury
- accelerated necrosis w/ massive influx of Ca - appear in <2 min after reperfusion - margins of infarcts b/w dead and viable tissue - > number in infarcts after reperfuse - --> sudden death - also caused by perioperative ischemia during cardiac surgery |
|
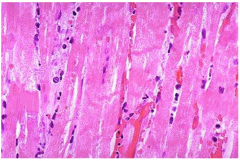
|
- contraction band necrosis - can't see the nuclei but can see striations
|
|
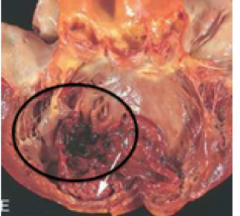
which complication of MI is this?
|
mural thrombus --> emboli that can break off and --> stroke
|
|
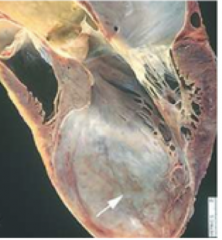
which complication of MI is this?
|
ventricular aneurysm
|
|
|
What are 2 causes of pericarditis after MI?
|
1) acute fibrinous pericarditis w/ transmural infarct: 2-3 days post-MI
2) post-MI syndrome (Dressler's) - autoimmune rxn - takes 1-8 weeks to develop |
|
|
whats the most common cause of sudden cardiac death?
|
- ischemic heart disease (found in 80-90% of those with SCD)
- 75% stenosis in >1 vessel, usually 90% in one |
|
|
what is graft ateriopathy?
|
- progressive thickening of intima w/ luminal narrowing
- limits long-term success of cardiac transplants - denervated heart does not allow patient to feel angina! |