![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
127 Cards in this Set
- Front
- Back
|
What is the capillary fragility test? |
Evaluates vascular integrity |
|
|
What are issues with using EDTA tubes for hemostasis testing? |
EDTA clumpers Platelet satellites Cold Plt antibodies |
|
|
What is IPF? What do low and high values indicate? |
Immature Platelet Fraction Measures number of immature platelets by presence of RNA (reticulated) More immature = more fluorescence Low to N IPF = decreased plt production Increased IPF = plt destruction |
|
|
Sodium citrate tube: What is the mechanism of anticoagulation? What CBC results can you report? |
Sodium citrate binds calcium CBC results: Can report WBC and PLTs -1:10 ratio needed -multiply by 1.1 for dilution correction |
|
|
What does the bleeding time test measure? |
Evaluates platelet function and number (OHSU ref range 2.5 - 9.5 minutes) |
|
|
What is the PFA-100 test? |
Measures closure time Replacement test for Bleeding time test -Collagen/ADP -Collagen/Epinephrine (Measures plt ability to adhere and aggregate under capillary flow conditions) |
|
|
What is the principle of aggregometry? |
Sample: PRP (PPP as a blank) Add agonist and measure light transmittance over time. As the platelets aggregate, the solution becomes less turbid and more light is transmitted over time Measures plt function and number |
|
|
What is primary aggregation and secondary aggregation in aggregometry? |
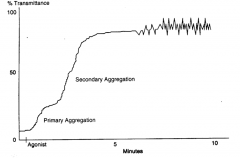
1* direct aggregation by agent (reversible) 2* aggregation mediated by release rxn |
|
|
What are the main platelet agonists used in aggregometry? |
–ADP: biphasic –Epinephrine/Norepinephrine: biphasic –Collagen: single phase with lag –Thrombin: biphasic –Arachidonic acid: biphasic –Ristocetin: biphasic Aggregation responses vary depending on agent and concentration (and patient...) |
|
|
What does Ristocetin measure? |
Measures plts ability to bind to vWF (The other agonists measure the plts ability to aggregate) |
|
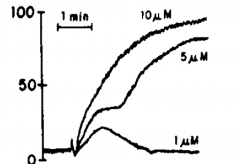
|
ADP 10um-High concentration 5um-Biphasic 1um-Low concentration so didn't stimulate enough release reaction for secondary aggregation to occur |
|
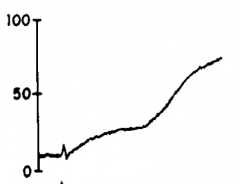
|
Epinephrine |
|
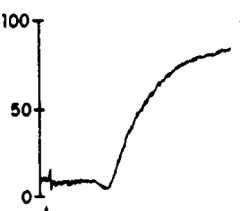
|
Collagen |
|
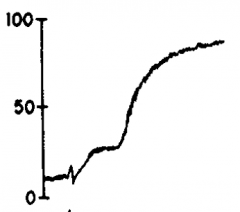
|
Thrombin |
|
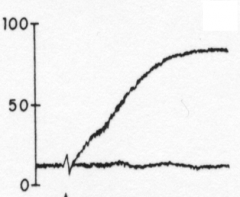
|
Arachidonic acid |
|
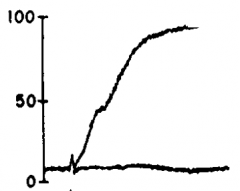
|
Ristocetin |
|
|
What are some factors affecting platelet aggregation measurement? |
pH (8.0 optimum) Temperature(370C) Stirring-constant Time- atleast 30 min Cuvetteandstir bar size / shape |
|
|
What are some patient factors that increase plt aggregation? |
exercise stress obesity highfat diet smoking diabetesmellitus |
|
|
What are some patient factors that decrease plt aggregation? |
alcohol aspirinand other anti-inflammatory drugs |
|
|
What is Hereditary Hemorrhagic Telangiectasia? |
Hereditary Vascular Disorder Single layer of endothelial cells = vessel fragility (NOT common) |
|
|
What is Ehlers-Danlos Syndrome? |
Hereditary Vascular Disorder (Also called Indian Rubberman) Collagen disorder that results in flexible joints and affects plt adhesion (NOT common) |
|
|
What is Allergic Purpura? |
Hereditary Vascular Disorder Autoimmune associated vascular injury Allergic manifestations (NOT common) |
|
|
What is Scurvy? |
Hereditary Vascular Disorder Vitamin C deficiency (Vitamin C is needed for collagen synthesis so results in decreased collagen = weak capillary walls) (NOT common) |
|
|
What is Bernard Soulier Syndrome? |
Disorder of Platelet Adhesion Lack of GP1b receptor on plts Lab Findings: Increased bleeding time, decreased plts, plt aggregation studies all normal except for with Ristocetin |
|
|
What is von Willebrands Disease Type I? |
Disorder of Platelet Adhesion Decrease in ALL multimers of vWF (Possibly due to decreased production or decreased release) Most common Type |
|
|
What is von Willebrand's Disease Type II (A-B, M, N)? |
Disorder of Platelet Adhesion Decrease in high MW multimers of vWF (Possibly due to inability to stabilize large ones) |
|
|
What is von Willebrand's Disease Type III? |
Disorder of Platelet Adhesion ALL multimers absent or severely decreased (Possibly due to decreased production or rapid breakdown at sites of synthesis) Most severe type! |
|
|
What is von Willebrand's Disease Plt Type? |
Disorder of Platelet Adhesion GP1b has increased affinity for vWF Platelets agglutinate spontaneously and then are removed = decreased plts and vWF |
|
|
What are the Lab findings in von Willebrand's Disease Type I? |
PT normal APTT increased Bleeding Time increased Plt aggregation studies all Normal except with Ristocetin Quantitate plasma vWF (ELISA, IEP, Latex particles) Separate multimers by MW (Gel Electrophoresis, IEP) |
|

|
11- Type 2A 10- Type 1 9- Type 2B 8- Normal |
|
|
What are the two tests to measure vWF activity? |
Ristocetin Induced Platelet Agglutination (RIPA) -Patient PRP in aggregometer -All responses normal except with Ristocetin -Measures ability of pt. plts to bind to vWF Ristocetin Cofactor (vWF:Rco) -Patient PPP and donor plts in aggregometer -Measures ability of pt. plasma vWF to bind to donor plts in presence of ristocetin |
|
|
What are five other problems that can affect platelet function? |
-Autoimmune disorders (IgG anti-plt) -Myeloproliferative disorders (malignancy decreases plt function) -Multiple Myeloma and Waldenstrom's (Ig coat plts and collagen) -Chronic Liver disease (direct toxic effects) -Drugs (modify plt membrane) |
|
|
What is Hereditary afibrinogenemia? |
Disorder of Aggregation Hereditary fibrinogen deficiency |
|
|
What is Uremia? |
Disorder of Aggregation Toxins interfere with plt function |
|
|
What is Glanzmann's Thrombathenia? |
Disorder of Aggregation Platelets lack GPIIb/IIIa complex Lab findings: Bleeding time- Increased, PT- N, APTT- N, Plt count- N, Plt aggregation studies all ABnormal except Normal with ristocetin |
|
|
What is the action of aspirin on plt function? |
Aspirin reduces plt aggregation -22% of patients taking aspirin become resistant -Resistance associated with thrombosis -Can detect with Plt aggregation studies (Arachidonic acid curve most sensitive) |
|
|
What are Storage Pool Diseases? |
Disorder of Release Reaction Platelets lack dense granules, which results in abnormal aggregation |
|
|
What is Hermansky-Pudlak syndrome? |
Disorder of Release Reaction (Also called "Swiss Cheese Plt Syndrome") Channels (canicular system) in plts are dilated, which results in deficient release reaction |
|
|
What is Gray Platelet Syndrome? |
Disorder of Release Reaction Marked decrease in alpha granules (Plts look agranular on smear b/c alpha granules are most abundant type) |
|
|
What is Wiskott Aldrich Syndrome? |
Disorder of Release Reaction (Also form of Congenital Hypoplasia) -Micro plts -Decreased dense + alpha granules -Decreased plts due to plt sequestration -B and T cell dysfunction = recurrent infections |
|
|
What is Chediak-Higashi Disorder? |
Disorder of Release Reaction Plts lack normal dense granules |
|
|
Aspirin Therapy can be associated with what type of disorder and what is the mechanism of action? |
Associated with Disorder of Release Reaction Inhibits Cyclooxygenase which is needed for TXA2 production |
|
|
What is Reactive Thrombocytosis? |
All is normal, body just reacting. Responsetoblood loss, major surgery, childbirth, tissue necrosis, inflammatorydisease,exercise, etc. |
|
|
What are the Myeloproliferative Disorders associated with Thrombocytosis? |
–Polycythemiavera (PV) –ChronicMyelocytic Leukemia(CML) –Primary Myelofibrosis –EssentialThrombocythemia (ET) |
|
|
What is May Hegglin Disorder? |
Decreased Platelet Production (Form of Congenital Hypoplasia) -Ineffective thrombopoiesis -Large/bizarre plts -Dohle-like bodies -Most patients are asymptomatic, may have bleeding and increased infections |
|
|
What is Neonatal Hypoplasia? |
Decreased Platelet Production Causes: -Newborns with Rubella- lack megakaryocytes -Drugs ingested during pregnancy- toxic to newborn megakaryocytes, will recover within weeks |
|
|
What are some of the causes of Acquired Hypoplasia? |
Decreased Platelet Production Irradiation, Drugs, Ethanol, Earlyaplasticanemia, Perniciousanemia, folatedeficiency, Viruses, Bacterialinfections, Malignancies, Myelodysplasticsyndromes Similar to causes of RBC hypoplasia |
|
|
What is Immune Thrombocytopenia Purpura (ITP) and what are the Lab Findings? |
Increased Platelet Destruction (Immune Mechanism) –PLTcount often< 20,000 –Pltlarge (variable size and shape) –BM- megakaryocytehyperplasia –Bleedingtime- increased –Deficientclot retraction |
|
|
What are the main characteristics of Chronic ITP? |
-Patient usually 20-50 yrs old -Fluctuating course -Bleeding episodes for days or weeks -Spontaneous remissions are uncommon -Early manifestation of AIDS |
|
|
What are the main characteristics of Acute ITP? |
-Patients usually kids -Often have history of viral infection (2-21 days prior) -Sometimes occurs after immunizations -Usually self-limiting -80% have spontaneous remission |
|
|
What are the possible mechanisms of Drug Induced Immune Effects and how does it affect platelets? |
Increased Platelet Destruction (Immune Mechanism) Mechanisms: -True auto-antibody develops -Ab forms after hapten-linkage drug to plt -Drug- antibody complex attaches to plt (Common Drugs: Heparin, Quinine, Quinidine) |
|
|
When should you suspect heparin induced immune effects? |
When a patient on heparin has a drastic 30-50% drop in PLT count |
|
|
What is HAT? |
Heparin-associated Thrombocytopenia Increased Platelet Destruction Direct NON-immune mediated plt activation Risk of bleeding, NOT associated with risk of thrombosis |
|
|
What is HIT? |
Heparin-Induced Thrombocytopenia (Type I) Increased Platelet Destruction (Immune Mechanism) Develops antibody to PF4-heparin complex Risk of bleeding, Low plt count |
|
|
What is HITTS? |
Heparin-Induced Thrombotic Thrombocytopenic Syndrome (Type II) Increased Platelet Destruction (Immune Mechanism) Develops antibody to PF4-heparin complex Risk of bleeding, VERY low plts, thrombosis |
|
|
What is the therapy for HIT and HITTS? |
Take patient off of heparin because auto-antibody is only active in presence of heparin Anticoagulant Substitutes: -Coumadin -Low MW heparin (may still cross-react) -Fondaparinux (Xa inhibitor) -Lepthirudin + Argatroban (thrombin inhibitors) |
|
|
How do you test for HIT and HITTS? |
HIPA Tests for presence of heparin-induced antibody in plasma Pateint PPP + donor plts + dilutions of heparin in aggregometer (if ab present, plts will aggregate) |
|
|
What is Neonatal Alloimmune Thrombocytopenia? |
Increased Platelet Destruction (Immune Mechanism) Mom alloantibody against baby plt antigen 1 in 5,000 newborns Pathophysiology same as HDN |
|
|
What is Neonatal Autoimmune Thrombocytopenia? |
Increased Platelet Destruction (Immune Mechanism) Mom has ITP or SLE so has autoantibody against own plts that cross-reacts with baby plts High Risk Delivery: Perform Fetal Scalp PLT count |
|
|
What is HELLP Syndrome? |
Hemolysis, ELevated Liver Enzymes, Low Plt Increased Platelet Destruction ("Non"-Immune Mechanism) Life threatening complication of pregnancy Preeclampsia/Eclampsia develop into HELLP |
|
|
What is the etiology of HELLP? |
-IntravascularPlateletactivation -Microvascular endothelialdamage -ThromboxaneA2release -Vasospasm -Vascularlesionsin multiple organs |
|
|
What is preeclampsia and eclampsia? |
Preeclampsia - Pregnancy inducedhypertension in association with either edema or proteinuria after 20 weeks gestation Eclampsia- Severe formof preeclampsia in which there are also seizures or coma |
|
|
What are the symptoms of HELLP? |
-Hypertension -Nausea -Malaise -Epigastric Pain -Rightupper quadrant tenderness -Edema -Cerebraland Visual disturbances -Proteinuria + Oliguria |
|
|
What are the Lab Findings of HELLP? |
Evidence of hemolysis (*schistocytes on smear) PLT count < 200,000/mm3 (low for pregnancy) Hepatic Dysfunction (elevated liver enzymes) Patient basically goes into DIC |
|
|
What is Thrombotic Thrombocytopenic Purpura? |
Increased Platelet Destruction -Auto-antibody against ADAMTS13 protease -Rare inherited ADAMTS13 deficiency Large, non-fragmented vWF occludes microvasculature (microthrombi) |
|
|
What are the lab findings in TTP and what is the treatment? |
-Hemolyticanemia with schistocytes -Thrombocytopenia(hemorrhage) -Fluctuatingneurological dysfunction -Fever -Progressiverenal disease Treatment- FFP, anti-plateletdrug (ex: asprin), steroidtreatment |
|
|
What is Hemolytic Uremic Syndrome? |
Increased Platelet Destruction Resembles TTP, typically found in kids 4-6 yrs -MAHA -Thrombocytopenia: sequestered in kidney, pltaggregates -Acuterenal failure -Absence of neurologic symptoms |
|
|
Why should a hemolyzed blood sample NOT be used for hemostasis testing? |
Because Erythrocetin released from RBCs has thromboplastic effect (will start clotting process) |
|
|
Not used EDTA or heparin tubes for coag tests? |
EDTA degrades Factor V Heparin inhibits coagulation factors |
|
|
What samples are generally used for platelet studies versus coag studies? |
Platelet studies use PRP Coag studies use PPP |
|
|
What are the components of the Intrinsic and Extrinsic Tenase complexes and what do they activate? |
Intrinsic: IXa, PF3, Ca2+, VIII(a) Extrinsic: TFIII, Ca2+, VIIa Tenase complexes activate factor X |
|
|
What are the components of the Prothrombinase complex and what does it activate? |
Prothrombinase complex: Xa, V, Ca2+, PF3 Activates prothrombin (II) to Thrombin (IIa) |
|
|
What factors are the "labile" factors? |
Factors V and VIII (Only factors that are not enzymes) |
|
|
What are the Magic Four? |
Factors II, VII, IX, X All vit K dependent (adds 2nd carboxyl group) Produced in the liver All depleted by oral anticoagulant therapy |
|
|
What is Factor I? |
Fibrinogen Ia = Fibrin monomer Acute phase reactant Made in the liver |
|
|
What is Factor II? |
Prothrombin IIa = Thrombin Thrombin responsible for activation of Factors V, VIII, XI, XIII, Protein C, TAFI, fibrinogen to fibrin Made in the liver, Vit K dependent |
|
|
What is TFIII? |
Tissue Factor III Found in all tissues Released from damaged tissue Cofactor of factor VII activation |
|
|
What is Factor IV? |
Ca2+ |
|
|
What is Factor V? |
Labile factor Made in the liver Activated by thrombin Inactivated by Protein C Works in Prothrombinase complex |
|
|
What is Factor VII? |
Proconvertin/Stable Factor Made in the liver Vit K dependent Activated by TFIII in presence of Ca2+ Works in Extrinsic Tenase complex ---> X Can also activate factor IX |
|
|
What is Factor VIII? |
vonWillebrand Factor Complex Produced in several tissues (mostly liver) Activated by thrombin Inactivated by Protein C Works in Intrinsic Tenase complex ---> X |
|
|
What is a Factor VIII deficiency? |
Hemophilia A
|
|
|
What is Factor IX? |
Christmas Factor Made in the liver Vit K dependent Activated by Factors XIa or VIIa Works in Intrinsic Tenase Complex ---> X |
|
|
What is a Factor IX deficiency? |
Hemophilia B |
|
|
What is Factor X? |
Stuart-Prower Made in the liver Vit K dependent Activated by Intrinsic and Extrinsic Tenases Works in prothrombinase complex ---> II |
|
|
What is Factor XI? |
Contact Factor Made in the liver Travels in blood with HMWK Activated by Factors XIIa, IIa Activates Factor IX |
|
|
What is a Factor XI deficiency? |
Hemophilia C |
|
|
What is Factor XII? |
Hageman Factor Made in the liver Contact factor Activated by contact with collagen Activates factor XI (with cofactor HMWK) and Prekallikren |
|
|
What does a factor XII, PK, or HMWK deficiency often cause? |
Thrombosis due to lack of fibrinolytic pathway activation |
|
|
What is Factor XIII? |
Fibrin Stabilizing Factor Made in the liver Activated by Thrombin in presence of Ca2+ Forms covalent bonds in D domains of polymerized fibrin |
|
|
What is PK? |
Prekallikren "Fletcher Factor" Made in the liver Contact factor Activated by Factor XIIa with cofactor HMWK Kallikren activates Factor XII and Plasminogen Kallikren hydrolyzes bradykinins from HMWK |
|
|
What is HMWK? |
High Molecular Weight Kininogen "Fitzgerald Facor" Made in the liver Contact factor Complexed with Factor XI and PK Cofactor with XIIa for activation of XI and PK Substrate for Kallikren ---> kinins |
|
|
What are the functions of Bradykinins? |
-Increasevascular permeability -Contractsmooth muscle -Dilatesmall blood vessels -Inducepain and inflammation -Releaseprostaglandins from tissues -Chemotaxis |
|
|
What is PF3? |
Platelet Factor 3 -Phospholipid that moves to outer surface of platelet when platelets are activated -Acts as supportive surface for coag cascade -Binding site for Vit K dependent factors -Works in Intrinsic Tenase and Prothrombinase complexes |
|
|
What is Protein C? |
Inactivates Va, VIIIa Liberates tissue plasminogen activator (TPA) Protein S is a cofactor Activated by Thrombin-Thrombomodulin complex (APC- activated form) Vit K dependent Made in the liver |
|
|
What is Thrombomodulin? |
Endothelial cell membrane glycoprotein Once thrombin bound, it can no longer activate fibrinogen ---> fibrin but can activate Protein C |
|
|
What is Protein S? |
Cofactor to Protein C Two forms: -Active: Free form that can participate in coagulation (40%) Vit K dependent, Made in liver, Non-proteolytic |
|
|
What is Protein Z? |
Major Role: Degrade factor Xa Cofactor to ZPI Requires Ca2+ and Phospholipids Slow acting on its own Vit K dependent (but no activation site, non-proteloytic), made in the liver |
|
|
What is ZPI? |
Protein Z-Related Protease Inhibitor Major Role: Degrade factor Xa Cofactor to Protein Z Action accelerated 1000X with Protein Z Also can degrade Factor XIa w/out Protein Z Vit K dependent, Made in the liver |
|
|
What is AT? |
Antithrombin Forms irreversible complexes with: IIa, IXa, Xa, XIa, XIIa, plasmin ---> to slowly neutralize these factors Made in the liver |
|
|
How does Heparin affect Antithrombin? |
Causes a conformational change in AT molecule which increases its inhibitory effect Heparin is not consumed in this process so it can dissociate and act on more AT molecules Heparin therapy results in decreased levels of circulating AT so need to ween patient off... |
|
|
What is Heparin-Cofactor II (HC-II)? |
Neutralizes Thrombin (IIa) Activity is accelerated in presence of heparin (but even still much slower activity than AT) Made in the liver |
|
|
What is Tissue Factor Pathway Inhibitor (TFPI)? |
Low concentration Inhibitor of Extrinsic pathway (TFIII/VIIa Tenase complex) Secreted into plasma by endothelial cells |
|
|
What do these collectively work to do? What would deficiencies in any most likely lead to? APC/Protein S, Protein Z, ZPI, AT, HC-II, TFPI |
These collectively work together to regulate coagulation Deficiency would most likely lead to thrombosis |
|
|
What are the endogenous and exogenous activators of plasminogen? |
Endogenous: -Factor XIIa, Kallikren -APC ---> TPA -Urokinase Exogenous: -Streptokinase -Urokinase -TPA |
|
|
What are the functions of plasmin? |
-Lyses fibrin and fibrinogen* -Inactivates factors Va and VIIIa -Degrades factor XIIa (inactivates) *(Plasmin usually localized within clot so will primarily break down fibrin. Any plasmin that escapes is destroyed by anti-plasmins) |
|
|
What are the end-products of primary and secondary fibrinolysis? |
Primary (fibrinogenolysis): 2 D fragments + 1 E fragment Secondary (fibrinolysis): 2 D fragments + 1 E fragment *D fragments differ. D fragments of fibrinogenolysis have small peptides A,B attached |
|
|
What are FDPs? |
Fibrinogen/Fibrin Degradation Products Can act as coagulation inhibitors because can bind to fibrin monomers preventing polymerization and clot formation |
|
|
D-dimers are present in primary or secondary fibrinolysis? |
Secondary fibrinolysis (fibrinolysis) Because fibrin monomers have factor XIIIa so when plasmin breaks down the fibrin clot it produces the D-dimers |
|
|
What is Alpha2-antiplasmin? |
Escapee Plasmin Patrol Binds and inactivates free circulating plasmin (Rapid inhibitor- prevents lysis of fibrinogen and degradation of factors V, VIII) Not very effective at inactivating plasmin bound to fibrin (which is good- not its function) |
|
|
What is Alpha2-Macroglobulin? |
Plasmin inhibitor ONLY when alpha2-antiplasmin binding sites are all saturated (Slower inhibitor- prevents lysis of fibrinogen and degradation of factors V, VIII) |
|
|
What are Plasminogen Activator Inhibitors (PAI-1 and PAI-2)? |
Neutralizes TPA and Urokinase Released from injured endothelial cells and activated platelets PAI-1 is most important inhibitor to the plasminogen activator system |
|
|
What is Thrombin Activatable Fibrinolytic Inhibitor (TAFI)? |
Inhibits/Slows fibrinolysis
Alters fibrin clot so it is less recognizable as substrate for plasmin Activated by Thrombin/Thrombomodulin complex |
|
|
What do Alpha2-antiplasmin, Alpha2-macroglobulin, PAI-1/2, and TAFI have in common? What would deficiencies probably lead to in the patient? |
All are regulators of Fibrinolysis/Fibrinogenolysis Deficiency would probably lead to bleeding |
|
|
What is a thrombosis and what is an embolis? |
Thrombosis is a pathological formation of a major clot(s) stuck at the site of formation Embolis is when a clot or chunk of a clot travels from the site where it was formed and can cause blockage elsewhere |
|
|
What are the principal clinical syndromes that result from thrombosis? |
-Acutemyocardial infarction (AMI) -Deep vein thrombosis (DVT) -Pulmonary embolism(PE) -Acute ischemic stroke -Acute peripheral arterial occlusion -Occlusionof indwelling catheters |
|
|
What is DIC? |
Acute episode when there is overwhelming stimulation of coagulation from extensive endothelial damage or release of tissue thromboplastin -Bleeding too much in one site (too much plasmin overwhelmes antiplasmins) -Clotting too much in another site (overwhelmed fibrinolysis) |
|
|
What are some causes of DIC? |
Bacterialinfections, intravascularparasites, surgery,majortissue trauma,castingof legs,burns,snakevenoms,AProL. Complicationsof pregnancy:deadfetus syndrome,amnioticfluid embolus (high levels TFIII),abruptio placentae,preeclampsia,eclampsia,HELLP,increasesin factors VII, VIII, IX, XII, I, decreasedProtein S, abortion. |
|
|
What are the clinical symptoms of DIC? |
-Oozing blood -Bleeding from multiple sites (due to consumption of clotting factors -Evidence of organ damage (due to fibrin deposition---decreased O2---tissue necrosis---release TFIII---perpetuate coagulation) |
|
|
What is the purpose of anti-coagulants or anti-thrombotics? |
Therapeutic purpose is to limit or prevent clotting The goal is to prevent thrombosis without causing hemorrhage |
|
|
Heparin Therapy: Use and Mechanism |
Use: Administered by IV, monitored by APTT or anti-Xa assay. **May cause HIT Mechanism: Heparincauses a conformational change in the AT (increases it’sinhibitory effect). AT slowly neutralizes factors IIa, IXa, Xa, XIa, XIIa, and plasmin |
|
|
Low Molecular Weight Heparin (LMWH) |
Has greater inhibitory effect on Xa
Administered subcutaneously, monitored by chromogenic anti-Xa assay (or not at all) Does not cause HIT but can cross react with HIT ab, so NOT a good Heparin sub in HIT pt. |
|
|
Warfarin (Trade name Coumadin) |
Long-Term oral antithrombotic Monitored MONTHLY by PT and INR Mechanism: Arrests vit K in storage form so body cannot make vit K dependent factors (II, VII, IX, X) or vit K dependent regulators |
|
|
What are Direct Thrombin Inhibitors (DTIs)? |
Anticoagulants that bind and neutralize thrombin (free and fibrin bound) Good substitute for heparin (esp. in HIT pt.) Monitored by APTT |
|
|
What are Direct Factor Xa Inhibitors?
|
Directly neutralize Factor Xa Monitored by Anti-Xa assay (ONLY in special circumstances) |
|
|
What is the mechanism of anti-platelet agents? |
Reduce platelet activation and function by one of the following: -Inhibiting cyclooxygenase to reduce TXA2 (aspirin) -Bind PLT membrane ADP receptor -Bind GPIIb/IIIa Monitored with PFA-100 |
|
|
What is the mechanism of thrombolytic agents in Thrombolytic Therapy? |
"Clot Busters" Serine proteases that convert plasminogen to plasmin for clot breakdown Two types: -Direct conversion of plasminogen to plasmin (almost always results in hypofibrinogenemia) -Stimulate limited plasminogen conversion where fibrin is not present (TPA) because has significant specificity for fibrin (less likelihood of fibrinogen breakdown too) |