![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
168 Cards in this Set
- Front
- Back
|
Plasma proteins are
|
Synthesized by the liver
|
|
|
Albumins
|
Carrier plasma proteins that help control plasma oncotic pressure (pulls/keeps fluid in the vessels)
|
|
|
Globulins
|
Carrier plasma proteins that.....need to update this card
|
|
|
Clotting factors
|
Plasma proteins, mainly fibrinogen that help form blood clots
|
|
|
Erythrocytes
|
Are responsible for tissue oxygenation. They are biconcave and have reversible deformity and live for 120 days before being recycled by the body.
|
|
|
Leukocytes
|
Are the white blood cells that defend the body against infection and remove debris. There are two categories of leukocytes: Agranulocytes and Granulocytes.
|
|
|
Granulocytes
|
A category of WBCs that have membrane bound granules in their cytoplasm capable of destroying microorganisms. They have both inflammatory and immune functions.
|
|
|
Agranulocytes
|
Monocytes: become macrophages in soft tissues
Lymphocytes: B, T, & NK cells |
|
|
Platelets
|
Disk shaped cytoplasmic fragments. They are formed by fragments of megakaryocytes. They're essential for blood coagulation and control of bleeding.
|
|
|
Peripheral Blood Smear
|
Slide from blood sample to assess size, shape, number of cells
|
|
|
Anemia
|
↓ in RBCs in blood or a ↓ quantity or quality of Hgb. Result from:
|
|
|
Leukopenia
|
A quantitative alteration of leukocytes that is characterized by an ↓ in number of white blood cells below the normal level of 4.5-11 x10⁹ L in adults. This is never normal
|
|
|
Thrombocytopenia
|
↓ in number of platelets
|
|
|
Poikilocytosis
|
RBCs have various shape abnormalities
|
|
|
Hypochromic
|
Pale RBC’s with ↓Hgb (lower MCHC value)
|
|
|
Microcytic
|
Smaller than normal RBC (lower MCV value)
|
|
|
Hyperchromic
|
Dense, dark RBC’s with ↑Hgb (higher MCHC value)
|
|
|
Macrocytic
|
Larger than normal RBC’s (higher MCV value)
|
|
|
Anisocytosis
|
RBCs have various sizes
|
|
|
Leukocytosis
|
A quantitative alteration of leukocytes that is characterized by an ↑ in number of WBC’s above the normal level of 4.5-11 x 10⁹ L in adults
|
|
|
Erythrocytosis
|
↑ in number of RBC’s
|
|
|
Neutrophilia (Granulocytosis)
|
Occurs either reactively or malignantly and is characterized by an ↑ number of neutrophils >3800 x 10⁶/L. Rarely, values can exceed 100,000/μL (typically indicates a myelocytic leukemia) which can ↑ blood viscosity to the point of thrombosis or vessel occlusion.
|
|
|
Neutrophil/PMN/Segs
|
Type of WBC that ↑ either reactively or malignantly. Represent 54-62% of WBCs (w/Bands 0-4%). Their life span is 6-12 hrs in circulation and 2-4 days in tissues. Because they are the first responders to infxn/inflammation, a large pool is held in venous sinuses of the marrow. Depletion of the stores of PMNs stimulates granulopoiesis to replenish.
|
|
|
Clinical Manifestations of Anemia (in general)
|
Reduced O2 carrying capacity leads to hypoxemia and hypoxia in the tissues. S/Sxs vary based on the body's compensatory mechanisms and include: syncope, angina, tachycardia, organ dysfunction. Classic anemia S/Sxs include: weakness, fatigue, pallor (skin, mucus membranes, conjunctiva), dyspnea, tachycardia.
|
|
|
Eosinophil
|
Type of WBC ↑ with allergies (Type 1 hypersensitivity), IgE/Extrinsic Asthma, chronic inflammation, and parasitic infection. They represent 1-3% of WBCs and live 4-5 hours. Stimulated by Cytokines, antigen-antibody complexes, and mast cell degranulation.
|
|
|
Basophil
|
Type of WBC ↑ in myeloprolipherative dz (CML, HL, polycythemia vera), can release heparin & histamine (if you subscribe to the theory that they are mast cells in the tissues). Represent <0.75% of WBCs and live hours - days.
|
|
|
Monocyte
|
Largest WBC in peripheral smear. Become macrophages once in the tissues and are responsible for sustained response to an infxn. More efficient than granulocytes in the control of mycobacterium & fungal infxn (less so w/bacteria).
|
|
|
Left–Shift/Leukemoid rxn
|
When demand for mature PMNs exceeds supply, the marrow begins releasing immature neutrophils (bands) into the blood. Diagrams of PMNs developing usually go from left to right so an increase in immature PMNs is a "left-shift". This rxn can also be seen in leukemia hence the term Leukemoid rxn.
|
|
|
Thalassemia Complications
|
MC complication is due to iron overload from transfusions (hemosiderosis), skin darkening from iron stimulated melanin production, sideroblastic cardiomyopathy (arrhythmias, CHF, recurrent pericarditis), hepatic fibrosis and cirrhosis (often by age 5), and a variety of endocrinological disorders. Death results from these complications but age varies based on # of transfusions required.
|
|
|
Platelets
|
Small, purplish anuclear cells – thrombocyte
|
|
|
Reticulocyte
|
Young RBC’s with residual RNA
|
|
|
↑ Reticulocytes
|
Normal bone marrow response to anemia
|
|
|
Alpha Thalassemia Syndromes
|
Four genes control the α globulin chain synthesis resulting in four forms of Alpha Thalassemia:
|
|
|
Pancytopenia
|
↓ in all 3 blood cell types– RBC’s, WBC’s and Plts
|
|
|
Thrombocytosis or Thrombocythemia
|
Quantitative abnormality characterized by ↑ number of platelets
|
|
|
Thrombocytopenia
|
|
|
|
Hematocrit
|
% of whole blood that is RBC’s
|
|
|
Iron Deficiency Anemia: Description & Etiology
|
MC type of anemia world-wide. Can result from ↓ dietary intake, excessive blood loss, or metabolic/functional iron deficiency or a combination of these. In the case of metabolic/functional iron deficiency iron stores may be sufficient but delivery to the bone marrow is inadequate or inadequately used in the marrow to maintain heme synthesis. H. pylori infxn can cause IDA as can menorrhagia in women. MC causes in men and women are:
|
|
|
Iron deficiency Anemia: Lab Findings
|
|
|
|
Iron Deficiency Anemia: 3 stages
|
IDA occurs when the demand for iron exceeds the supply an develops slowly over three overlapping stages.
|
|
|
Iron Deficiency Anemia: S/Sxs
|
Primary s/sxs are general anemia s/sxs of weakness, fatigue, tachycardia, palpitations, dyspnea on exertion. As the IDA progresses additional s/sxs including chelosis, glossitis, brittle nails, koilonychia, hyposalivation, dysphagia, esophageal webs (Plummer-Vinson Syndrome), irritability, headache, parasthesia, vasomotor disturbances, confusion, memory loss, disorientation, permanent cognitive impairment in children, pica can also be a S/Sx as well as a cause
|
|
|
Sickle Cell Disease: Description
|
A group of d/os characterized by the presence of an abnormal form of Hgb (Hgb-S). Abnormal Hgb-S is formed as the result of a single point mutation where a valine replaces a glutamic acid. The result is that Hgb-S reacts to deoxygenation and dehydration by solidifying and stretching the RBC into an elongated or sickle shaped cell that gets stuck in the microvasculature.
|
|
|
Sickle Cell Disease: Genetics
|
SCD is an inherited autosomal recessive disorder with a single substitution of a valine for a glutamic acid. The homozygous form of this is what is known as Sickle Cell Disease and is the most severe form. The heterozygous form results in Sickle Cell Trait and rarely has clinical manifestations. This dz is MC in people of equatorial Africa, Near East, Mediterranean, & Indian descent. It is believed to have some evolutionary advantages against malaria.
|
|
|
Sickle Cell Disease: S/Sxs
|
Hemolytic anemia that is characterized by acute, painful episodes of vaso-occlusion (attacks) that result in dactylitis, acute pain, splenic sequestration. As the attack continues it results in more tissue hypoxia and hypoxemia which triggers even more sickling. Long term effects include multi–organ system failure due to vaso–occlusion.
|
|
|
Sickle Cell Disease: Treatment
|
REFER to hematologist. Supplement w/folic acid (as in all chronic anemias), exchange transfusions during crisis, hydroxurea long term. Stem cell transplant before multi-organ system damage can cure >80% of children, but HLA matched donors are RARE. Stem cell transplant in adults is still under investigation. During an attack/crisis need to keep hydrated and oxygenated as well as manage pain (is severe).
|
|
|
Sickle Cell Disease: Effects on Spleen
|
Hyposplenism (Howell-Jolly bodies)or functional asplenia leads to infection
|
|
|
Sickle Cell Disease: Lab results
|
↓ HCT (20-30%), ↑ reticulocytes, 5-50% of RBCs are irreversibly sickled, Howell–Jolly bodies from hyposplenism (HJ bodies unique to this dz), Target cells, ↑ WBCs 12,000-15,000/μL, reactive thrombocytosis may occur, ↑ indirect bilirubin levels. Dx is confirmed w/Hemoglobin electrophoresis (GOLD STANDARD). Hgb-S is typically 85-98% of all Hgb.
|
|
|
Alpha Thalassemia Trait: S/Sxs
|
Mildest form of alpha thalessemia. These ppl are typically asymptomatic having at the most a very mild microcytosis. This is the carrier state with a single abnormal α allele.
|
|
|
Thalassemia
|
Inherited autosomal recessive disorders of that cause an impaired rate of synthesis of Alpha & Beta globulin chains. Either can be characterized as major (homozygous gene), intermedia, or minor (heterozygous gene). Results in a microcytic-hypochromic hemolytic anemia that can be mild or serious enough to cause death in utero (hydrops fetalis).
|
|
|
Beta Thalassemia Syndromes
|
Defect resulting in an uncoupling of the α & β chain synthesis. Two genes control the β chain synthesis and depression of this synthesis results in RBCs having ↓ Hgb and accumulations of free α chains which are unstable and precipitate in the cell. Erythroblasts w/precipitates are destroyed by phagocytes in the marrow resulting in ineffective EPOiesis & anemia. Some of the erythroblasts do survive and mature but are then destroyed in the spleen (mild hemolytic anemia). β Thalassemias are MC than the α Thalassemias.
|
|
|
Alpha Thalassemia Minor
|
A mild, microcytic-hypochromic reticulocytosis that is virtually identical to Beta Thalassemia Minor. Bone marrow hyperplasia, ↑ serum Fe concentrations, and splenomegaly are moderate. These ppl have 2 abnormal α globulin chain alleles.
|
|
|
Beta Thalassemia Major (Cooley Anemia) – S/Sxs
|
2 abnormal β globulin chains (thalassemic alleles). Symptoms present in the first year of life and resulting anemia is severely microcytic-hypochromic and results in significant cardiovascular burden and high output CHF. Hepatosplenomegaly is common as is cholelithiasis and icterus. Significant hyperplasia of bone marrow in the long bones, skull/facial bones, and spine, although spinal impairment retards growth and subsequent upper and lower limb discrepancy. Require lifelong transfusions that may result in iron overload.
|
|
|
Beta Thalassemia Intermedia: S/Sxs
|
2 abnormal β globulin chains (thalassemic alleles) but results in a milder chronic hemolytic anemia that do not require transfusions except under periods of stress or during aplastic crises. Will still develop hepatosplenomegaly and bony deformities. Iron overload is also a risk from transfusions.
|
|
|
Beta Thalassemia Minor: S/Sxs
|
1 normal β globulin chain & 1 abnormal β globulin chain (thalassemic allele). Typically causes a mild to moderate microcytic-hypochromic anemia, mild splenomegaly, bronze skin tone, reticulocytosis varies w/degree of severity of anemia and then correlates w/hyperplasia of bone marrow. These patients are usually ASYMPTOMATIC.
|
|
|
Thalassemia: Dx & Labs
|
|
|
|
Anemia Compensatory Mechanisms: Cardiovascular
|
↓ in blood volume due to loss of cells results in ↑ extracellular fluid from tissues into blood stream to ↑ blood volume. At the same time HR ↑, SV ↑, & ↑ vasodilation. Leads to hyperdynamic circulation which results in cardiac murmurs & ↑ cardiac output which eventually results in cardiac failure
|
|
|
Hemolytic Anemias
|
|
|
|
Hemolytic Anemia Dx
|
↑ Retic count (reticulocytosis), ↓HCT, and metabolic byproducts of hemolysis: ↑indirect bilirubin, LDH, ↓haptoglobin
|
|
|
Aplastic Anemia: Management
|
|
|
|
Thalassemia: Tx/Management
|
|
|
|
Aplastic Anemia
|
↓ or absent hematopoetic precursors in marrow = pancytopenia. Results from injury to or suppression of the stem cells (chemo, sulfa drugs, phenotoin, radiation, autoimmune d/os like SLE, or inherited d/os like Fanconi anemia). Patients may present w/general anemia s/sxs; s/sxs of leukopenia like infxn; s/sxs of thrombocytopenia like purpura & petechiae. SHOULDN'T HAVE: bone pain, hepatosplenomegaly, or lymphadenopathy (those indicate cancer).
|
|
|
Aplastic Anemia: Lab results
|
|
|
|
Thalassemia Epidemiology
|
|
|
|
Alpha Thalassemia: Hemoglobin H Disease S/Sxs
|
Similar to Beta Thalassemia Major but less severe. This is a chronic hemolytic anemia of variable severity. It is microcytic-hypochromic anemia with bone marrow hyperplasia & hepatosplenomegaly evident. Rarely require transfusion except in times of crisis or stress. These individuals have 3 abnormal α globulin chain alleles.
|
|
|
Acute lymphoblastic leukemia
|
Most common malignancy in children
|
|
|
Anemia Compensatory Mechanisms: O2 demands & renal response |
↑ O2 demands for the work of the heart results in angina. Kidneys sense ↓ O2 and ↑ EPO to stimulate bone marrow to ↑ RBC production. If the anemia is acute or severe kidneys sense a ↓ in blood flow which ↑ the renin-aldosterone response resulting in ↑ Salt & H2O retention which ↑ extracellular fluid uptake into the blood to ↑ blood volume. |
|
|
Anemia SEFxs: Neuro & GI |
If the anemia is due to B12 deficiency, myelin degeneration can occur causing a loss of nerve fibers in the spinal cord resulting in parasthesia, ataxia, extreme weakness, spasticity, and reflex abnormalities. ↓ O2 to the GI system can result in abdominal pain, N/V & anorexia. |
|
|
Normochromic-Macrocytic Anemia (in general) |
Also called megaloblastic anemia due to the large size of the stem cells produced in the marrow. Caused by defective DNA synthesis resulting from B12 or folate deficiency which are the coenzymes used in the nuclear maturation & DNA synthesis pathways. RNA & Hgb production is unaffected resulting in an RBC that has unequal growth of the nucleus and the cytoplasm (abnormally large RBC w/small nucleus). These defective RBCs die prematurely causing the anemia. |
|
|
Pernicious Anemia Description & Etiology |
Most common type of macrocytic anemia afflicting people > 30 yo but taking 20-30 yrs to develop which is why the median age of dx is 60 (esp. white women >35). MCly caused by a lack of intrinsic factor (IF) from the gastric parietal cells. IF is the transporter required for B12 absorption in the ileum. Can be congenital, surgically induced, or an autoimmune response. The autoimmune response destroys gastric parietal cells preventing most IF from being secreted and also destroys any IF that is secreted preventing IF-B12 complexes from forming resulting in B12 deficiency. H. pylori infxn can be the beginning of the autoimmune response that destroys parietal cells. |
|
|
Pernicious Anemia: S/Sxs |
Initial s/sxs are vague and nonspecific. Once dz has progressed to the point where Hgb is <8 g/dL then weakness, fatigue, parasthesia of the feet and fingers, ataxia, ↓ sense of position & vibration, ↓ memory, loss of appetite, abdominal pain, weight loss, sore tongue that is beefy red and smooth 2ry to atrophic glossitis, sallow skin (excess bilirubin/ictarus), and/or pallor. Hepatomegaly indicates right sided heart failure may present in the elderly along with splenomegaly which is nonpalpable. Once neuro S/Sxs present they are irreversible regardless of Tx! |
|
|
Pernicious Anemia: Risk Factors/Underlying causes |
|
|
|
Pernicious Anemia: Lab values |
|
|
|
Pernicious Anemia: Tx |
Parenteral B12 replacement (100 mcg) is needed daily for the first week, weekly for the first month, then monthly for life. Once stores have been replenished, Pt could use oral or sublingual B12 if desired as there is some passive diffusion even in the absence of IF. Serum B12 levels should be monitored to ensure okay. Pts are typically also folate deficient so concurrent supplementation recommended. |
|
|
Folate Deficiency Anemia: Description/Etiology |
Folate is an essential vitamin/coenzyme required for DNA/RNA synthesis w/in the maturing erythrocyte. Specifically it is required for the synthesis of thymine, which impacts cells undergoing rapid division. Folate is not reliant on another mechanism for absorption into the small intestine. Folate deficiency is more common than B12 deficiency. Impaired DNA synthesis results in both megaloblastic cells and apoptosis of erythroblasts in the late stages of erythropoiesis, thus leading to anemia. |
|
|
Folate Deficiency Anemia: S/Sxs |
Cachectic/malnourished appearance, cheilosis, stomatitis, painful ulcerations of the buccal mucosa & tongue (burning mouth syndrome), dysphagia, flatulence, watery diarrhea, changed to GI tract similar to sprue. Generally neuro s/sxs not seen unless due to B12 deficiency which often accompanies folate deficiency. |
|
|
Folate Deficiency Anemia: Risk Factors |
|
|
|
Folate Deficiency Anemia: Lab values |
RBC folate level: < 150 ng/mL |
|
|
Folate Deficiency Anemia: Tx |
Oral supplementation of folate is required until the liver stores are built up again, 1mg/day is sufficient for most ETOHs may require 5mg/day. For pregnant/lactating women a prophylactic dose of 0.1-0.4 mg/day is given. |
|
|
Microcytic-hypochromic Anemias (in general) |
Characterized by abnormally small erythrocytes with ↓ Hgb, even normal RBCs have ↓ Hgb. Can result from:
Specific disorders are Iron deficiency anemia, Sideroblastic anemia, Thalassemias |
|
|
Iron Deficiency Anemia: Tx |
Oral supplementation is the primary method of treatment. Typically Ferrous Sulfate 325 mg TID on an empty stomach. Compliance is poor due to GI upset. Therapy should be continued for 3-6 months after initial anemia is corrected to build up stores again. Parenteral iron is available for patients who cannot tolerate or cannot absorb the oral preparation. |
|
|
Normocytic-normochromic anemias |
Characterized by cells that are normal in size and Hgb content but are insufficient in number due to hemolysis that is either intravascular or extravascular. Two types are autoimmune hemolytic anemia and drug induced hemolytic anemia. |
|
|
Autoimmune Hemolytic Anemias |
An extrinsic, acquired disorders caused by autoantibodies against antigens normally on the surface of erythrocytes. There are three types: warm reactive antibody type, cold agglutinin type and cold hemolysin type. |
|
|
Warm Autoimmune hemolytic anemia |
Uncommon disorder but the most common type of AIHA, representing 80-90% of all AIHA cases. Occurs in individuals > 40 yo, and approx 1/2 are secondary to other dz like lymphoma, chronic lymphocytic leukemia, other neoplastic disorders, or SLE. Caused by IgG antibodies binding to erythrocytes at normal body temp which then bind to the Fc receptors on monocytes and splenic macrophages, resulting in removal of the RBC membrane which forms a spherocyte. The spherocytes cannot pass through the splenic sinusoids and get trapped in the red pulp of the spleen. Hemolysis occurs in the spleen (extravascular). |
|
|
Cold Agglutinin autoimmune hemolytic anemia |
A less common AIHA that affects mostly mddle-aged and older adults. IgM antibodies bind to erythrocytes at colder temperatures. May appear acutely during recovery from infxn w/mono, mycoplasma pneumoniae, disseminated TB or may occur in conjunction w/lymphoid neoplasm. Anemia may be severe but is self-limiting. The IgM activates opsonization by C3b resulting in rapid phagocytosis in the liver (Kupfer cells). If the level of IgM is high, hemagglutination may occur in the capillaries of exposed sites (fingers, toes, ears). Prolonged exposure to cold may result in gangrene. |
|
|
Drug Induced Hemolytic anemia |
Occurs when an allergic rxn against foreign antigens (typically low molecular weight drugs) binds to proteins on the surface of erythrocytes. IgG antibodies against the drug or the unique antigen formed by the drug + protein bind to the erythrocyte at normal body temps. Sometimes the drug, IgM/IgG antibodies and C3b form complexes that then bind to C3b receptors on erythrocytes. In one instance methyldopa provoked a true AIHA by triggering anitbodies to adhere to an erythrocyte. All of the above result hemolysis in the liver and spleen. |
|
|
AIHA: Warm Type S/Sxs |
Variable severity anemia that typically has rapid onset that may be life threatening. Fatigue, dyspnea, angina pectoris, heart failure are all possible. Physical exam usually reveals icterus and splenomegaly (due to trapped spherocytes), abdominal pain, fever, coca cola urine or urine with "ground coffee" appearance. |
|
|
AIHA: Warm Type Lab results |
Severe cases may show a Hct < 10% (normal is 36-41). ↑ reticulocytes and spherocytes seen on smear. In severe cases nucleated RBCs may also be present. ↑ serum indirect bilirubin, ↓ haptoglobin. Dx is made using Coombs test (direct or indirect). Direct antiglobulin test (coombs test) is positive in 90% of all cases. Indirect antiglobulin (coombs) test may or may not be positive. |
|
|
AIHA: Warm Type Tx |
Refer to hematologist. Initial tx can include oral prednisone to suppress immune system and slow the rate of agglutination/marking. If CS are ineffective, splenectomy may be indicated. Additional treatments may include Rituximab, Danazol, or other immunosuppresive agents. |
|
|
AIHA: Cold Agglutinin Type S/Sxs |
Exposure to cold often results in mottled or numb fingers or toes, acrocyanosis, episodic low back pain, dark colored urine. |
|
|
AIH: Cold Agglutinin Type Lab results |
Mild anemia with ↑ reticulocytes and very rarely spherocytes. Smear at room temp shows agglutinated RBCs. Direct antiglobulin (Coombs) test is positive for complement only. Serum cold agglutinin titer will be positive. Serum protein electrophoresis may reveal IgM (M for mittens when cold). Indirect bilirubin ↑ and haptoglobin ↓. |
|
|
AIH: Cold Agglutinin Type Tx |
Largely symptomatic and involves avoiding colder temps. Splenectomy and prednisone are rarely effective, (except when associated with lymphoproliferative d/o) because hemolysis occurs in the liver and blood stream. Rituximab is the tx of choice. Severe dz requires referral to hematologist. |
|
|
Neutropenia |
↓ in number of circulating PMNs. Clinical dx when < 1,800/μL. If < 1,000/μL emergent referral. If < 500/μL life threatening risk of infxn is present. It can be genetic (congenital or cyclic) or acquired and can be the result of decreased/ineffective production or increased destruction, abnormal distribution/sequestration secondary to other disorders. |
|
|
Neutrophilia: Causes |
Surgery, burns, MI, RA, bacterial infxn, fungal infxn, acute inflammation, hypoxia, exercise, emotional stress, hemorrhage, long term CS use, epi use, heparin use, DM acidosis, eclampsia, Gout, thyroid storm, polycythemia vera, malignancies of Liver, GI tract, or marrow (lymphocytes, CML). |
|
|
Neutropenia: Genetic/Congenital Causes |
Include cyclic and congenital immunodeficiency dzs and syndromes like: Kostmann, Shwachman-Diamond, Diamond-Blackfan, etc. |
|
|
Neutropenia: Acquired causes leading to ↓ neutrophil production |
|
|
|
Neutropenia: Acquired causes leading to ↑ neutrophil destruction or abnormal distribution/sequestration |
|
|
|
Eosinophilia |
When Eosinophils > 200/μL (200 x 10⁶ L) in peripheral blood. MC cause is due to allergies (hay fever, atopic dermatitis, eczema, Extrinsic asthma) & drug hypersensitivity (streptomycin, PCN, propranolol). Additional causes include parasitic infxn, chronic infxns (TB, leprosy, fungal), autoimmune, and a variety of malignancies. |
|
|
Idiopathic Hypereosinophilic Syndrome |
A group of disorders characterized by double or triple the normal count of eosinophils. Results in diffuse tissue damage of endocardium, CNS, lungs, and spleen. Tx: is CS or hydroxurea. Prognosis is poor if not aggressively treated (only 10% survive 3 years). |
|
|
Basophilia |
When basophils >40/μL (40 x 10⁶ L) in the blood. Causes include: Infxn (measles, chickenpox), chronic inflammation (RA), myeloproliferative dz (CML, HL, polycythemia vera) |
|
|
Monocytosis |
When monocytes > 300/μL (300 x 10⁶ L) in the blood. Often is during the recovery phase of neutropenia associated w/bacterial infxn (compensatory mechanism). Can also occur w/SBE and in chronic infxns (EBV: mono, Syphilis, TB, autoimmune dz). Also correlates to the amount of damage after MI. MC cause is malignancy (Myelogenous leukemia, hodgkin's lymph) |
|
|
Lymphocytes |
These are the B (memory & AB production), T (memory, helper, cytotoxic killer), and NK cells (cancer & virus killers) that are involved in the innate AND adaptive immune systems. Long lifespans. |
|
|
Lymphocytosis |
When the lymphocyte count is >2,500/μL (2,500 x 10⁶ L). MC cause is acute viral infxn, particularly EBV: mono infxn (also w/CMV, RSV, Hepatitis). Also seen in chronic syphilis infxn & Leukemia. |
|
|
Anemia of Chronic Dz (in general) |
A mild - moderate normocytic or microcytic anemia resulting from ↓ erythropoiesis in ppl w/chronic systemic dz or inflammation. One of the MC causes of anemia probably only secondary to IDA in overall incidence. Elderly are commonly affected since they are most likely to have both chronic dz and long-term effects of inflammation. |
|
|
Anemia of Chronic Dz: Causes |
|
|
|
Anemia of Chronic Dz: How it happens |
Chronic dz/inflammation releases a large variety of cytokines (TNF-α, IFN-γ, IL-β, IL-3,6). ↑ hepcidin results in ↓ Ferroportin resulting in ↓ Fe release from macrophages. Release of Lactoferrin & Apoferrin by neutrophils in chronic inflamm also binds Fe making it less avail for EPOiesis (altered Fe metab). Often kidney dz accompanies chronic dz and results in ↓ EPO release which also ↓ EPOiesis. |
|
|
Anemia of Chronic Dz: Clinical Manifestations |
S/Sxs of ACD are typically those of the underlying condition. If the anemia becomes significant enough to produce it's own S/Sxs (↓ Hgb), then they typically mirror IDA |
|
|
Anemia of Chronic Dz: Lab results |
Hct rarely ↓ below 60% of pts baseline except in Chronic Kid dz. MCV is typically normocytic or slightly microcytic. RBC morphology is typically normal. Reticulocyte count is typically normal or slightly ↓. Serum Fe & transferrin levels are typically ↓ with a trasnferrin sat extremely ↓ (leading to incorrect dx: IDA). |
|
|
Anemia of Chronic Dz: Tx |
Treat the underlying cause/chronic dz. If severe, may need to give parenteral recombinant EPO and/or a transfusion of RBCs |
|
|
Polycythemia (relative) |
Hemoconcentration of the blood associated with dehydration caused by ↓ H2O intake, diarrhea, excessive vomiting, ↑ use of diuretics. Usually self limiting and resolves with resolution of underlying cause. |
|
|
Polycythemia (absolute) |
Consists of two forms:
|
|
|
Polycythemia Vera (primary): How it works |
Part of a group of d/os collectively called chronic myeloproliferative d/os. PV is rare but white males >60 yo are MCly afflicted. It is a chronic neoplastic, nonmalignant condition of overproduction of RBCs (commonly WBCs & Plts too) and splenomegaly. >95% of ppl w/PV have a defect in the JAK2 gene that essentially causes RBC precursor cells to always be receptive to EPO. |
|
|
Polycythemia Vera (primary): Clinical manifestations |
Abdominal pain, palpable splenomegaly, headache, dizziness, tinnitus, blurred vision (retinal engorgement), fatigue, GENERALIZED PRURITIS AFTER EXPOSURE TO HEAT OR WATER, epistaxis, thrombosis (stroke, retinal occlusion, spleen), HTN, hepatomegaly. |
|
|
Polycythemia Vera (primary): Lab results |
|
|
|
Polycythemia Vera (primary): Tx |
Weekly phlebotomy to remove blood until Hct is back to a normal level (men: 41%, women: 36%). DO NOT give Fe supplement - already overloaded on iron. Symptomatic tx of complications like thrombosis. May require referral to hematologist for marrow suppression. |
|
|
Infectious Mononucleosis |
Mono is an acute, self-limiting, neoplastic lymphoproliferative clinical syndrome characterized by acute viral infxn of B cells w/EBV. B cells have receptors for EBV resulting in infxn. Lymphocytosis results from unaffected B cells producing IgG, IgM, IgA while a simultaneous proliferation of cytotoxic T cells takes place. Cellular proliferation in the lymphoid tissues results in lymphadenopathy, tonsilomegaly, splenomegaly, and occassionally hepatomegaly. |
|
|
Acute Leukemia |
Characterized by undifferentiated or immature cells, usually blast cells, IN THE BONE MARROW and the onset of dz is abrupt and rapid w/ a short survival time. The two types of acute leukemia are: acute lymphocytic (ALL) and acute myelogenous (AML) |
|
|
Chronic Leukemia |
Characterized by more differentiated/mature cells IN THE BONE MARROW that do not fxn properly. It has a relatively slow progression. The two types of chronic leukemia are: chronic lymphocytic (CLL) and chronic myelogenous (CML). |
|
|
Philadelphia chromosome |
Translocation between chromosome 9 & 22 results in this BCR-ABL1 or Philadelphia chromosome and is the most common genetic link in people w/leukemia. BCR-ABL1 appears to excessively activate intracellular pathways, leading to increased proliferation, decreased sensitivity to apoptosis, and premature release of immature cells into circulation. |
|
|
Leukemia (in general) |
Described as clonal disorders in that a single progenitor cell undergoes a malignant transformation. The subsequent malignant blasts crowd out the marrow and cause cellular proliferation of the other lines to cease (pancytopenia). |
|
|
Acute Lymphocytic Leukemia: Description & Statistics |
ALL is a progressive neoplastic dz characterized by the presence of >30% lymphoblasts in the marrow or blood. 80% of ALL cases occur in children, and it is the MC leukemia in children (most often occurring before age 10). Median age of dx is 13 yo and children have a 78% survival rate. Adults represent 20% of ALL cases but have a significantly higher mortality rate (30% for ppl >20 dropping to 15% for ppl > 60 and 5% for ppl > 70). |
|
|
Acute Lymphocytic Leukemia: Genetics/Pathophys |
Approx 75% of ALL cases in children originate from precursor B cells. Adult ALL is a mix of precursor B & precursor T cells. A small percentage are neither B nor T cells and are called "null cell". Specific phenotypes vary based on their progression through the cells lines. Trisomy 21 children have an ↑ risk of ALL & AML. Ppl w/other genetic conditions also have an ↑ risk of ALL. |
|
|
Acute Myelogenous Leukemia: Pathophys & statisics |
Results from an abnormal proliferation of myeloid precursor cells, ↓ in apoptosis, and an arrest in cellular differentiation, resulting in marrow/periph smears w/leukocytosis and predominance of blast cells. As blasts ↑, normal myelocytic cells, megakaryocytes, and erythrocytes are replaced resulting in the S/Sxs. AML is the MC adult leukemia with the median dx age of 67. Approx 60-70% of adults with AML will attain complete remission status, but remission rates are inversely related to age. |
|
|
Acute Myelogenous Leukemia: Genetics & Risk Factors |
AML ↑ w/age, exposure to radiation, benzene, & chemotherapy. Genetic conditions like Trisomy 21, Fanconi aplastic anemia, & congenital X-linked agammaglobulinemia all have ↑ risk of AML. Phenotypes are classified based on their progression through the cell line at the time of Dx. |
|
|
Acute Leukemia: Clinical manifestations |
S/Sxs of ALL/AML are similar and progress rapidly (days or weeks) including: fatigue caused by anemia, bleeding from thrombocytopenia, pyrexia due to infxn (esp by GNR). Bleeding can manifest as purpura, petechiae, ecchymosis, gingival bleeding, hematuria, menorrhagia, metrorrhagia, discolored skin, DIC, anorexia and cachexia, dysphagia and ↓ sensitivity to sour/sweet tastes, abdominal, breast, & bone/joint pain. CNS S/Sxs include papilledema, headache, vomiting, facial palsy, visual & auditory disturbances. Hepatosplenomegaly & lymphadenopathy occur more frequently in ALL than AML. |
|
|
Acute Leukemia: Lab results |
Infxn results from neutropenia (500/μL or 500 x 10⁹ L). A dramatic presentation may include hyperleukocytosis or a total WBC > 100,000/μL (results in many of the CNS s/sxs due to vascular traffic jam). The hallmark is pancytopenia + circulating blasts (smear may be ablastic). Bone marrow is typically hypercellular and dominated by blasts (>20% blasts required to make dx). ALL pts may have mediastinal mass on CXR. The Auer rod is pathognomonic of AML and secures the Dx. |
|
|
Acute Leukemia: Tx |
All Pts referred to a heme/onc specialist. Pts up to age 60 are treated with the goal of cure. The first step is chemotherapy to obtain complete remission. The type of chemo varies based on the type (including phenotype) of leukemia. Additional tx may include allogenic stem cell transplantation. |
|
|
Chronic Leukemia: Description and statistics |
Prognosis is better since these are a slower progressing dz. Chronic leukemias account for the majority of cases in adults and 30% of leukemias in the western world. Incidences of CML and CLL increase significantly in ppl >40 w/prevalence in 6-8th decades. |
|
|
Chronic Leukemia: CLL Pathophys |
Malignant transformation and progressive accumulation of monoclonal B lymphocytes that are partially mature and have not yet encountered antigens. Rarely it can be of T cell origin (<5%). Cells accumulate in the marrow but do not interfere w/other cell line production. Accumulation is the result of cell cycle arrest (G₀/G₁ phase) and ↓ sensitivity to apoptosis. Failure of these B cells to mature into plasma cells results in hypogammaglobulinemia (60% of ppl). |
|
|
Chronic Leukemia: CLL lab results |
Hallmark is isolated lymphocytosis. WBC >20,000/μL all the way up to >100,000/μL. 75-98% of circulating cells are lymphocytes. Hct & Plt count are typically normal at onset. Marrow may be 30% lymphocytes and normocellular or hypercellular. Hypogammaglobulinemia is present in 50% of pts. |
|
|
Chronic Leukemia: CML Pathophys |
A member of the myeloproliferative d/o that also includes PV. CML is clonal, arising from a hematopoietic stem cell. CML cells are heterogenous in differentiation depending on stage of dz. Philadelphia chromosome present in >95% of cases. Accumulation of additional mutations leads to more aggressive dz. |
|
|
Chronic Leukemia: CLL clinical manifestations |
70% of ppl w/CLL are asymptomatic at the time of Dx. MC s/sx is lymphadenopathy. Most significant effect of CLL is suppression of humoral immunity and ↑ infxn w/encapsulated MOs. Neutropenia common ↑ infxn risk. Lymph nodes, liver, spleen, salivary glands are MC site of infiltration. 10% of ppl develop more aggressive form and experience fever, night sweats, fatigue, ↑LD, hypercalcemia, anemia, thrombocytopenia. |
|
|
Chronic Leukemia: CML clinical manifestations |
|
|
|
Chronic Leukemia: CML Lab results |
Characterized by ↑ WBC at Dx of 150,000/μL (150 x 10⁹ L). Periph blood shows a left shift w/mature forms dominating. Blasts typically <5%, basophilia & eosinophilia may be present. At Dx Pt usually NOT anemic and RBC morphology is normal. Plts are normal or slightly ↑. Marrow is hypercellular, w/left-shifted myelopoiesis (myeloblasts <5%). Blast phase accelerates and shows ↑ blasts (>20% of marrow), ↑ anemia & thrombocytopenia HALLMARK IS DETECTION OF bcr/abl GENE USING PCR. |
|
|
Chronic Leukemia: Tx |
Varies based on CML vs CLL...this card to up updated after adult med lecture! |
|
|
Malignant Lymphoma |
A diverse group of neoplasms that develop from the proliferation of malignant lymphocytes in the lymphoid system including primary lymphoid tissue (thymus, bone marrow) and secondary lymphoid tissue (lymph nodes, spleen, tonsils, intestinal lymphoid tissue - MALT). Divided into two major types: Hodgkin's and Non-Hodgkin's. |
|
|
Hodgkin Lymphoma: Description & Statistics |
A malignant lymphoma first described by Thomas Hodgkin in 1832. Incidence is slightly ↑ in men vs women and the median age of Dx is 64. Incidence rates have ↓ due to improved Dx accuracy. Occurs Whites > African Americans & Denmark, Netherlands & US have ↑ level. HL peaks in ppl in 20-30's or later in the 60-70's.
|
|
|
Hodgkin Lymphoma: Pathyphys |
Characterized by its orderly progression from one group of lymph nodes to another, the development of systemic S/Sxs, and presence of Reed-Sternberg cells. RS cells represent the malignant lymphocytes, are large and bi-nucleate that cells secrete/release cytokines (IL-10, TGFB-β) resulting in accumulation of inflammatory cells that produce local and systemic effects. RS cells are necessary for the Dx of HL but not specific to HL. Etiology is unknown but it is suggested that EBV infxn is linked to malignant mutation/transformation of B cells in germinal line. |
|
|
Hodgkin Lymphoma: Clinical Manifestations |
Painless lymphadenopathy in the cervical, axillary, inguinal, and retroperitoneal lymph nodes, asymptomatic mediastinal mass on CXR, splenomegaly, abdominal mass. Constitional S/Sxs may include: intermittent fever, night sweats, pruritus, fatigue. Constitutional S/Sxs combined with weight loss have poor prognosis. |
|
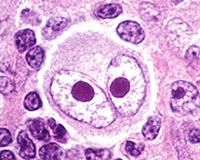
What is shown in this picture? |
Reed-Sternberg cell that is diagnostic for HL. |
|
|
Hodgkins Lymphoma: Lab results |
Pts are often asymptomatic for several years. CXR, lymphangiography, biopsy is recommended for ppl w/fever of unknown origin AND peripheral lymphadenopathy. Biopsy results showing Reed-Sternberg cells and a cellular infiltrate is HIGHLY indicative of HL. Thrombocytosis, leukocytosis, eosinophilia, ↑ ESR, ↑ alkaline phosphatase. |
|
|
Hodgkins Lymphoma: Tx |
Chemo & ABVD remains the standard first line tx for HL. All Pts should be treated w/curative intent. Prognosis in advanced stage dz depends on seven features: stage, age, gender, hemoglobin, albumin, WBC, lymphocyte count. Cure rate = 75% if 2 features present & 55% when >3 features present. |
|
|
Non-Hodgkins Lymphoma: Description & Statistics |
A generic term for a diverse group of neoplasms (B cell, T cell, NK cell). These malignancies are different from HL in that they do NOT have RS cells. Median age of Dx is 67 and the incidence has more than doubled in the last 40 years. |
|
|
Non-Hodgkins Lymphoma: Pathophys |
NHL is ultimately a progressive clonal expansion of B/T/NK cells. Genetic lesions (many environmentally induced) affect proto-oncogenes and tumor-suppressor genes resulting in imortality and proliferation of malignant cells. B cells account for > 80-90% of NHLs. NHL tumors are classified based on origin, differentiation, and rate of proliferation. |
|
|
Non-Hodgkins Lymphoma: Clinical Manifestations |
Begin as localized or generalized lymphadenopathy in the cervical, axillary, inguinal, & femoral chains. The lymphadenopathy is typically painless and transforms/enlarges over many years. Early stage, low-grade lymphoma is also called indolent due to slow progression. Night sweats, fever, extranodular involvement, cytopenia (marrow involvement), hepatosplenomegaly, fatigue & weakness prevalent in more advanced stages (intermediate or aggressive). |
|
|
Non-Hodgkins Lymphoma: Lab results |
Peripheral blood is typically normal even w/extensive marrow involvement. Marrow involvement is manifested as paratrabecular monoclonal lymphoid aggregates. Dx made by tissue biopsy of lymph node or possible needle aspiration. |
|
|
Non-Hodgkins Lymphoma: Tx |
Diverse and depends on stage, type (B or T cell), histologic status, symptoms, age, and comorbidities. Tx ranges from careful observation, to chemotherapy, to radiation therapy, to stem cell transplant, to monoclonal antibody (rituximab). |
|
|
Burkitt Lymphoma: Description |
It is the fastest growing, highly aggressive non-hodgkin lymphoma that grows primarily in the jaw and facial bones. Accounts for 30% of childhood lymphomas world-wide. MC in children from East Africa and New Guinea. Rare in the US and typically involves the abdomen. |
|
|
Burkitt Lymphoma: Pathophys |
EBV is associated w/90% of all cases. B cells have surface receptors for EBV which leads to chromosomal translocations that result in an overexpression of the C-MYC proto-oncogene causing uncontrolled cell growth. |
|
|
Burkitt Lymphoma: S/Sxs & Tx |
In non-African BL, MC symptom is abdominal swelling and may involve other organs. Presents w/type B symptoms (night sweats, weight loss, fever). African BL is treated using radiotherapy & cyclophosphamide. Non-African BL is more resistant to tx but Adjuvant tx w/monoclonal antibodies (rituximab) is promising. |
|
|
Thrombocytopenia: Causes |
Results from ↓ plt production, ↑ plt consumption or both. The condition can be congenital or acquired and primary or secondary to other acquired conditions. The MC cause of thrombocytopenia are the result of ↑ platlet consumption (ITP, TTP, HIT). |
|
|
Immune Thrombocytopenia Purpura (formerly Idiopathic Thrombocytopenia Purpura): Acute |
MC cause of thrombocytopenia secondary to ↑ platlet destruction. Can be acute or chronic. Acute is typical in children, lasts 1-2 months then self resolves. Acute ITP is usually secondary to infxn (esp viral), SLE, or drug allergies (PCN). Under these conditions antigen forms immune complexes w/Abs (IgG) which then bind to the Fc portion (glycoprotein) on the platelets leading to their destruction in the spleen. |
|
|
Immune Thrombocytopenia Purpura (formerly Idiopathic Thrombocytopenia Purpura): Chronic |
Chronic form associated with autoantibodies against platelet specific antigens. MC in adults w/↑ prevalence in women 20-40 yo. Chronic form tends to get worse |
|
|
Immune Thrombocytopenia Purpura (formerly Idiopathic Thrombocytopenia Purpura): S/Sxs |
Minor bleeding, petechiae, purpura over the course of several days that progress to major hemorrhage from mucosal sites (epistaxis, hematuria, menorrhagia, bleeding gums). Typically spontaneous bleeding/hemorrhage doesn't occur until platelets are <20,000-30,000/μL. |
|
|
Immune Thrombocytopenia Purpura (formerly Idiopathic Thrombocytopenia Purpura): Tx |
Only ppl with platlets below the 30,000 mark or those with significant bleeding should be treated. Tx includes CS w/ or w/out IVIG or WinRho (anti-D). Transfusions may be given if active bleeding present. |
|
|
Thrombotic Thrombocytopenic Purpura |
Characterized by thrombotic microangiopathy in which platelets aggregate and cause occlusion of the arterioles and capillaries in microcirculation. May lead to organ ischemia esp kidney, brain, heart. Two types: Familial (chronic, relapsing, seen in kids) & Acquired Idiopathic (acute, severe, MC women in 30s). Related to dysfxn of plasma metalloprotease responsible for digesting VonWillebrand Factor. |
|
|
Thrombotic Thrombocytopenic Purpura: Pentad of Findings |
Thrombocytopenia: typically severe Fever Renal Insufficiency Neurologic Impairment Microangiopathic hemolytic anemia: ischemia & vessel occlusion |
|
|
Disseminated Intravascular Coagulation (DIC) |
Acquired clinical syndrome characterized by the intravascular activation of coagulation w/loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfxn. It is clotting and hemorrhaging at the same time. |
|
|
Disseminated Intravascular Coagulation (DIC): Causes & progression |
|
|
|
Disseminated Intravascular Coagulation (DIC): Summary |
Amount of activated thrombin exceeds the body's antithrombins and the thrombin does not remain localized. Widespread thromboses created, cause widespread ischemia, infarction, and organ hypoperfusion. |
|
|
Disseminated Intravascular Coagulation (DIC): Clinical Manifestations & Lab Results |
Bleeding at multiple sites (Incisions, IV access & cath access) and may be widespread (purpura fulminans). Early in DIC plts & fibrinogen counts are normal or slightly ↓ from baseline. Progressive thrombocytopenia and prolonged activated partial thromboplastin time (aPTT) and prothrombin time (PT), and ↓ levels of fibrinogen. D-dimer levels are ↑ due to the activation of coagulation and diffuse cross-linking of fibrin. Schistocytes appear in the smear due to shearing of RBCs through the microvasculature. |
|
|
Disseminated Intravascular Coagulation (DIC): Tx |
Underlying cause of the DIC must be treated with Abx, chemo, surgery, or delivery of baby. If clinically significant bleeding occurs then hemostasis must be achieved. Transfusion is indicated only if clinically significant hemorrhage has occurred. |
|
|
Hemolytic Dz of the Newborn/Erythroblastosis fetalis: Description |
Acquired congenital hemolytic anemia where maternal and baby blood are antigenically incompatible (either ABO incompatible or if baby is Rh+ and mother is Rh-), causing the mother's immune system to produce antibodies against baby's erythrocytes. Once the baby's erythrocytes are tagged w/antibodies, they are phagocytozed in the baby's spleen. |
|
|
Hemolytic Dz of the Newborn/Erythroblastosis fetalis: Statistics |
ABO incompatibility occurs in about 20-25% of pregnancies but only 1 in 10 result in HDN. Rh incompatibility occurs in <10% of all pregnancies and rarely causes HDN in the first pregnancy as it is that pregnancy that results in the mother's immune system developing Abs against the Rh factor. Subsequent pregnancies that are Rh incompatible are at risk and even then only 5% result in HDN. Only 1:3 HDN cases are Rh incompatibility the other 2:3 are ABO incompatibility. |
|
|
Hemolytic Dz of the Newborn/Erythroblastosis fetalis: Pathophys |
Once the Abs are formed against the baby's RBCs, IgG coats the offending cells and tags them for destruction in the baby's spleen. As hemolysis progresses the baby become more anemic and EPOiesis accelerrates and reticulocytes ↑ in circulation. The unconjuctated/indirect bilirubin crosses the placenta and is excreted by mom. Once baby is born hyperbilirubinemia occurs as there is no longer placental excretion. Pathophys of HDN is worse w/Rh incompatibility and is more likely to result in life-threatening anemia or death in utero or permanent damage to the baby's CNS. Babies that die in utero are typically still born and have gross edema body-wide, a condition called, Hydrops Fetalis. |
|
|
Glucose 6 phosphate dehydrogenase (G6PD) deficiency |
An inherited, X-linked, recessive disorder that is most fully expressed in homozygous males (partial expression and carrier states are possible in heterozygous females). Most common in Tropical & Subtropical zones of the Eastern Hemisphere (evolutionary advantage against malaria). G6PD is an enzyme that allows RBCs to maintain metabolic processes despite injury due to oxidative stress. |
|
|
Glucose 6 phosphate dehydrogenase (G6PD) deficiency: pathophys |
Presence of certain drugs (sulfa, antimalarials, salicylates, napthaquinolones), ingestion of fava beans, hypoxemia, infxn, fever, or acidosis can all cause injury/oxidative stress to the RBC. G6PD deficiency is typically asymptomatic unless these conditions occur. Intense or prolonged exposure to these conditions triggers Hgb denaturation and formation of Heinz bodies which results in RBC membrane damage and removal in the spleen (extravascular hemolysis). This will cease when these conditions are removed. |
|
|
Glucose 6 phosphate dehydrogenase (G6PD) deficiency: Tx |
Treatment is determined based on clinical syndrome. Avoiding or ceasing exposure to the trigger is important for affected individuals as well as pregnant or nursing heterozygous women. Affected individuals should be supplemented with folic acid. |
|
|
Heparin Induced Thrombocytopenia |
Platelet Factor 4 binds to heparin resulting in a conformational change of a heparin PF4 complex. IgG against the heparin-PF4 complex which binds to the complex which then binds to the Fc-gamma portion of the platelet which then activates it to release more PF4 furthering the coagulation cascade. Once all the factors and platelets are spent, similar to DIC, thrombocytopenia occurs and bleeding begins (5-10 days later |