Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
198 Cards in this Set
- Front
- Back
|
Where are WBCs distributed?
|
- Bone marrow
- Peripheral blood - Lymph nodes, thymus, spleen, tonsils, adenoids, Peyer patches - Mucosa-Associated Lymphoid Tissue (MALT) in lung and GI tract |
|
|
Where does WBC maturation begin? With what?
|
- Starts in bone marrow
- With Pluripotent Stem Cell |
|
|
What lineages can the Pluripotent Hematopoietic Stem Cell in BM differentiate into?
|
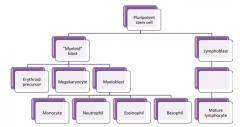
- Myeloid
- Erythroid - Megarkaryocytic - Lymphoid |
|
|
What are the types of Myeloid WBCs?
|
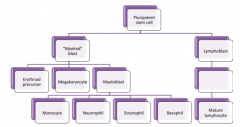
- Monocytes
- Neutrophils - Eosinophils - Basophils |
|
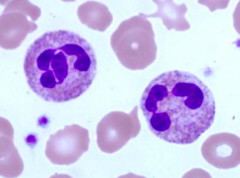
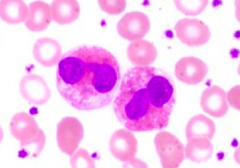
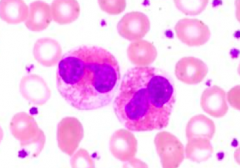
What kind of cells are these?
|
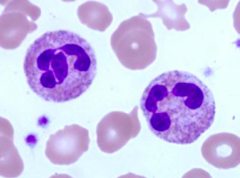
Neutrophils
|
|
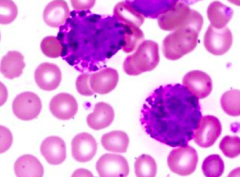
What kind of cells are these?
|
Basophils
|
|
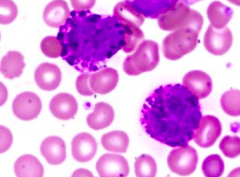
What kind of cells are these?
|
Eosinophils
|
|
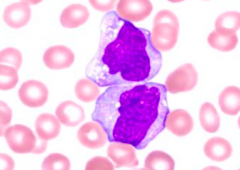
What kind of cells are these?
|
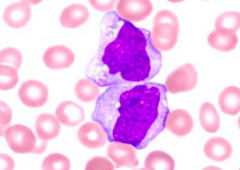
Monocytes
|
|
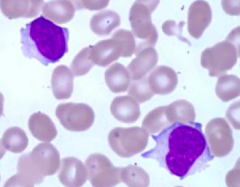
What kind of cells are these?
|
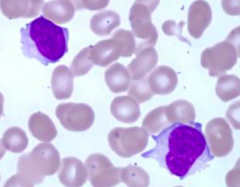
Lymphocytes
|
|
|
What is the term for NON-neoplastic conditions affecting WBCs?
|
Benign Leukocyte Disorders (clonal disorder)
|
|
|
What are the types of Neoplastic conditions affecting WBCs? Location?
|
- Leukemias: proliferation of neoplastic cells, primarily in BM and PB
- Lymphomas: proliferation of neoplastic cells, primarily in Lymph Nodes and extramedullary lymphoid tissue |
|
|
What are the types of Benign Leukocyte Disorders?
|
- Qualitative (structural or functional) disorders
- Quantitative disorders (increased WBCs = cytoses; decreased WBCs = cytopenias) |
|
|
What is the term for a Benign Leukocyte Disorder with increased WBC number?
|
Cytoses
|
|
|
What is the term for a Benign Leukocyte Disorder with decreased WBC number?
|
Cytopenia
|
|
|
What are the types of Quantitative Benign Leukocyte Disorders?
|
- Cytoses (increased WBCs)
- Cytopenias (decreased WBCs) - Leukemoid Reaction - Leukoerythroblastic Reaction |
|
|
What happens in a Leukemoid Reaction?
|
- Benign, exaggerated response to infection
- NOT leukemia |
|
|
What are the blood cell counts in Leukemoid Reaction?
|
- ↑ Leukocytes > 50,000 / µL
- May involve neutrophils, lymphocytes, or eosinophils |
|
|
What change in WBCs will a severe infection in perforating appendicitis cause?
|
Neutrophilic Leukemoid Reaction:
- Benign Leukocyte Disorder - Absolute leukocyte count usually >50,000/µL - Specifically elevated neutrophils |
|
|
What change in WBCs will whopping cough cause?
|
Lymphocytic Leukemoid Reaction:
- Benign Leukocyte Disorder - Absolute leukocyte count usually >50,000/µL - Specifically elevated lymphocytes |
|
|
What change in WBCs will a nematode infestation, such as cutaneous larva migrans cause?
|
Eosinophilic Leukemoid Reaction:
- Benign Leukocyte Disorder - Absolute leukocyte count usually >50,000/µL - Specifically elevated eosinophils |
|
|
What are the types of Leukemoid Reactions? Causes?
|
- Neutrophilic: severe infection in perforating appendicitis
- Lymphocytic: whooping cough (Bordetella pertussis) - Eosinophilic: nematode infestation (eg, cutaneous larva migrans) |
|
|
What happens in a Leukoerythroblastic Reaction?
|
- Benign Leukocyte Disorder
- Presence of immature bone marrow cells in the peripheral blood - Immature WBCs (blasts) or RBCs (nucleated RBCs) |
|
|
What can cause a Leukoerythroblastic Reaction?
|
- Bone marrow infiltrative process such as bone marrow fibrosis or metastatic cancer
- A severe bone marrow stress d/t benign conditions such as sepsis or growth factor administration may also cause a similar phenomenon - Leads to immature WBCs (blasts) or RBCs (nucleated) to be released into the peripheral blood |
|
|
What is the blood count in a Neutrophilia? What can cause a Neutrophilia?
|
- Absolute Neutrophil count > 7000 / µL
- Infection (eg, acute appendicitis) - Sterile inflammation w/ necrosis (eg, acute MI) - Drugs (eg, granulocyte colony stimulating factor (G-CSF), steroids, catecholamines, lithium) |
|
|
What drugs can cause Neutrophilia?
|
- G-CSF (granulocyte colony stimulating factor)
- Steroids - Catecholamines - Lithium |
|
|
What happens to neutrophils during an infection?
|
- Increased neutrophils = neutrophilia
- Neutrophils are not only increased in number, but also have prominent blue primary granules (instead of usual orange secondary granules) = Toxic Change |
|
|
What happens during a "toxic change" to neutrophils?
|
During infection, prominent neutrophil granules go from orange secondary granules to blue primary granules
|
|
|
What is the blood count in a Neutropenia? What can cause a Neutropenia?
|
- Absolute neutrophil count < 1500 / µL
- Aplastic Anemia - Immune destruction (eg, systemic lupus erythematous) - Septic shock - Chemotherapy for various malignancies |
|
|
What is the blood count in a Eosinophilia? What can cause a Eosinophilia?
|
- Absolute eosinophil count > 700 / µL
- Type I hypersensitivity rxn (eg, bronchial asthma, penicillin allergy, hay fever) - Invasive helminths (eg, strongiloidiasis, hookworm) - Hypocortisolism (eg, Addison's disease) - Neoplasms (eg, Hodgkin's lymphoma) |
|
|
What is the blood count in a Basophilia? What can cause a Basophilia?
|
- Absolute basophil count > 200 / µL
- Chronic Kidney Disease * Chronic Myelogenous Leukemia (and other myeloproliferative neoplasms) |
|
|
What blood count abnormality is most frequently encountered in Chronic Myelogenous Leukemia?
|
Basophilia ( > 200 / µL )
|
|
|
What are the types of Myeloid Neoplasms?
|
- Myeloproliferative Neoplasms (MPNs)
- Myelodysplastic Syndromes (MDS) - Acute Myeloid Leukemia (AML) |
|
|
What type of disorders are Myeloproliferative Neoplasms (MPNs) and Myelodysplastic Syndromes (MDS)? What kind of cells are affected?
|
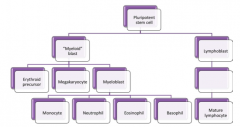
- Myeloid neoplasms
- Neoplastic stem cell disorders - May involve one or more specific cell lineages |
|
|
What kind of data is necessary to diagnose a Myeloproliferative Neoplasm (MPN) or a Myelodysplastic Syndrome (MDS)?
|
Combination of:
- Morphologic features - Immunophenotypic features - Genetic features - Clinical features * With rare exceptions, in order to make a diagnosis we need to synthesize information from various sources |
|
|
What characterizes Myeloproliferative Neoplasms (MPNs)?
|
- Clonal hematopoietic stem cell disorders
* Proliferation of one or more of myeloid lineages (granulocytic, erythroid, megakaryocytic, or mast cells) - Early precursor cell is affected, but one specific cell lineage may be primarily affected downstream |
|
|
What are the most common types of Myeloproliferative Neoplasms (MPNs)? What cell lineage is involved?
|
- Chronic Myelogenous Leukemia, BCR-ABL+ (CML) = neutrophils or monocytes
- Polycythemia Vera (PV) = RBCs - Primary Myelofibrosis (PMF) = neutrophils or monocytes - Essential Thrombocythemia (ET) = platelets |
|
|
Which Myeloproliferative Neoplasms (MPNs) cause proliferation of neutrophils and monocytes?
|
- Chronic Myelogenous Leukemia, BCR-ABL positive (CML)
- Primary Myelofibrosis (PMF) |
|
|
Which Myeloproliferative Neoplasms (MPNs) cause proliferation of red blood cells?
|
Polycythemia Vera (PV)
|
|
|
Which Myeloproliferative Neoplasms (MPNs) cause proliferation of platelets?
|
Essential Thrombocythemia (ET)
|
|
|
When do Myeloproliferative Neoplasms (MPNs) more commonly occur? How common?
|
- Adults: 40s-60s
- Relatively rare: 6-10 / 100,000 / year |
|
|
What are the commonalities of the Myeloproliferative Neoplasms (MPNs)?
|
* Hypercellular BM
* Effective hematopoiesis → ↑ numbers of peripheral blood myeloid derivatives (WBCs, RBCs, platelets) = Cytoses - Splenomegaly and/or hepatomegaly - Potential for disease progression to either BM fibrosis or acute leukemia |
|
|
What can the Myeloproliferative Neoplasms (MPNs) progress to?
|
- Bone Marrow Fibrosis
- Acute Leukemia |
|
|
What kind of genetic/molecular abnormality do the Myeloproliferative Neoplasms (MPNs) have?
|
- Cytogenetic or molecular abnormalities
- Leads to expression of protein w/ increased tyrosine kinase activity |
|
|
What disorders are commonly affected by increased tyrosine kinase activity?
|
Myeloproliferative Neoplasms (MPNs):
- Chronic myelogenous leukemia (CML) - Polycythemia vera (PV) - Primary myelofibrosis (PMF) - Essential thrombocythemia (ET) |
|
|
What kind of state can you consider Myeloproliferative Neoplasms (MPNs) and Myelodysplastic Syndromes (MDS)?
|
Pre-Acute Leukemic state (neoplastic condition)
|
|
|
What is the overall effect of Myeloproliferative Neoplasms (MPNs)?
|
Increased Cell Proliferation
|
|
|
What are the primary features of the Myeloproliferative Neoplasms (MPNs)?
|
* Hypercellular bone marrow
* Ineffective hematopoiesis → ↓ peripheral blood counts = Cytopenias |
|
|
What is the overall effect of Myelodypslastic Syndromes (MDS)?
|
Increased Cell Death
|
|
|
What are the most significant differences between Myeloproliferative Neoplasms (MPNs) and Myelodysplastic Syndromes (MDS)?
|
- MPN: hypercellular BM + ↑ PB counts (↑ cell proliferation)
- MDS: hypercellular BM + ↓ PB counts (↑ cell death) |
|
|
What kind of disease is Chronic Myelogenous Leukemia? What genetic/molecular change occurs?
|
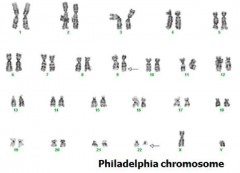
- Type of Myeloproliferative Neoplasms (MPNs)
* BCR-ABL fusion gene → produces a protein w/ Tyrosine Kinase activity * t(9;22) → Philadelphia Chromosome → BCR-ABL fusion gene |
|
|
What symptoms do patients with Chronic Myelogenous Leukemia have?
|
Non-specific symptoms:
- Fatigue - Weakness - Weight loss - Anorexia - Hepatosplenomegaly |
|
|
What are the peripheral blood cell count changes in Chronic Myelogenous Leukemia?
|
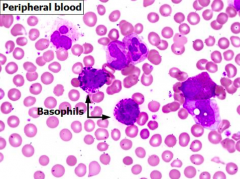
- ↑↑↑ Leukocytosis (>100,000 / µL)
- Neutrophilia (>7000 / µL) - Immature myeloid cells, rare myeloblasts (2-3%) * Basophilia - Thrombocytosis (50% of cases) |
|
|
What are the bone marrow cell changes in Chronic Myelogenous Leukemia?
|
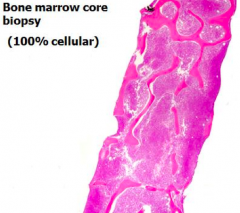
- Hypercellular (~100%)
- Granulocytic hyperplasia (predominance of the precursors that form neutrophils, basophils, and eosinophils) |
|
|
What gene change results from a t(9;22) translocation? Implications? What disease?
|
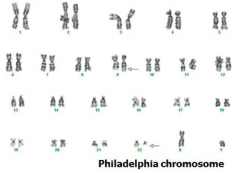
BCR-ABL fusion gene → Philadelphia chromosome → ↑ tyrosine kinase activity
(Chronic Myelogenous Leukemia) |
|
|
How do you detect the BCR-ABL translocation t(9;22)? Diagnostic of?
|
- FISH, rtPCR
- NOT specific for Chronic Myelogenous Leukemia, ALSO seen in Acute Leukemia (both myeloid and lymphoblastic) = CML, AML, ALL |
|
|
What are the stages of Chronic Myelogenous Leukemia? Length of time?
|
1. Chronic Phase - 3 years
2. Accelerated Phase - 1 year 3. Blast Phase (equivalent to AML or ALL, >20% blasts in PB or BM) |
|
|
How do you treat (not cure) Chronic Myelogenous Leukemia? Response?
|
Administer BCR-ABL tyrosine kinase inhibitors:
*Imatinib mesylate (Gleevac)* - 85% have good response, but this does NOT cure, so must be taken continuously for life - Also some other tyrosine kinase inhibitors |
|
|
How can you potentially cure Chronic Myelogenous Leukemia? Response?
|
Allogeneic stem cell transplant (only for patients with a matched donor that can tolerate this procedure) - possibly a cure
|
|
|
Which Myeloproliferative Neoplasm is associated with increased RBC mass? Mutation? Implications?
|
Polycythemia Vera (PV):
- Janus 2 Kinase (JAK2) gene (tyrosine kinase) mutation involved in signal transduction pathways → constitutively active - Leads to increased proliferation of RBC precursors |
|
|
What are the symptoms of Polycythemia Vera (PV)?
|
- Splenomegaly
- Thrombotic events d/t hyperviscosity (eg, hepatic vein thrombosis) - Gout (↑ cell breakdown → ↑ uric acid) - Ruddy face, pruritus after bathing, or peptic ulcer disease (d/t ↑ Histamine released from mast cells) |
|
|
What are the lab findings of Polycythemia Vera (PV)?
|
- ↑ RBC mass → ↑ [Hb] and/or ↑ RBC count
- Leukocytosis - Thrombocytosis - ↓ Serum [Epo] in presence of normal SaO2 |
|
|
How do the levels of Epo help in diagnosing Polycythemia Vera (PV) compared to other conditions that may cause erythrocytosis (increased RBC mass)?
|
- PV: ↓ serum [Epo]
- Other conditions causing erythrocytosis may be d/t exogenous Epo production or d/t ↑ Epo production secondary to ↓SaO2 |
|
|
Polycythemia Vera:
- RBC Mass? - Plasma Volume? - SaO2? - Epo? |
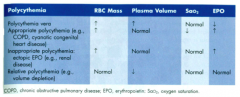
- ↑ RBC mass
- ↑ Plasma volume - Normal SaO2 - ↓ Epo |
|
|
Polycythemia / Erythrocytosis d/t COPD, cyanotic congenital heart disease:
- RBC Mass? - Plasma Volume? - SaO2? - Epo? |
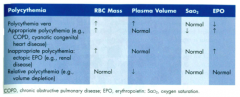
- ↑ RBC mass
- Normal Plasma volume - ↓ SaO2 - ↑ Epo |
|
|
Polycythemia / Erythrocytosis d/t Ectopic Epo (eg, renal disease):
- RBC Mass? - Plasma Volume? - SaO2? - Epo? |
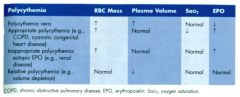
- ↑ RBC mass
- Normal plasma volume - Normal SaO2 - ↑ Epo |
|
|
Relative Polycythemia / Erythrocytosis d/t volume depletion:
- RBC Mass? - Plasma Volume? - SaO2? - Epo? |
- Normal RBC mass
- ↓ Plasma volume - Normal SaO2 - Normal Epo |
|
|
What are the diagnostic criteria for Polycythemia Vera?
|
- JAK2 mutation (↑ tyrosine kinase activity)
- Hypercellular BM w/ Erythroid Hyperplasia |
|
|
What is the clinical course and prognosis of Polycythemia Vera?
|
- Median survival > 10 years
- Most patients die of thrombosis or hemorrhage |
|
|
How do you treat Polycythemia Vera?
|
Managed w/ conservative treatment (eg, phlebotomy)
|
|
|
What can Polycythemia Vera progress to? How often?
|
- 15-20% evolve to "spent phase" which is similar to primary myelofibrosis
- 2-3% develop MDS or AML |
|
|
What are the characteristics of Primary Myelofibrosis (PMF)? What kind of disease is it?
|
- Rapid development of BM fibrosis and extramedullary hematpoiesis (EMH) in spleen, liver, and lymph nodes
- Myeloproliferative Neoplasm |
|
|
What are the symptoms of Primary Myelofibrosis (PMF)?
|
Splenomegaly → Portal HTN
|
|
|
What happens in the early and typical stages of Primary Myelofibrosis (PMF)?
|
- Early: pre-fibrotic stage shows peripheral blood leukocytosis
- Typical: fibrotic stage shows peripheral blood normochromic, normocytic anemia w/ frequent teardrop cells, and a leukoerythroblastic reaction |
|
|
What happens early on in Primary Myelofibrosis (PMF)?
|
- Pre-fibrotic stage
- Peripheral blood leukocytosis |
|
|
What happens later in Primary Myelofibrosis (PMF)?
|
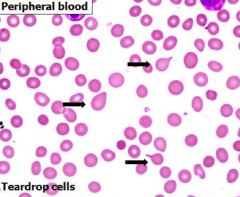
- Typical / fibrotic stage
- Peripheral blood w/ normochromic, normocytic anemia - Frequent teardrop cells - Leukoerythroblastic reaction |
|
|
What does a BM biopsy in Primary Myelofibrosis (PMF) show?
|
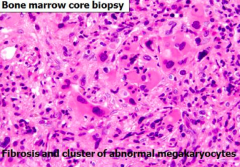
- Fibrosis
- Clusters of atypical megakaryocytes - Generally increased platelet count, may be variable |
|
|
What genetic/molecular abnormality is associated with Primary Myelofibrosis (PMF)?
|
JAK2 mutation found in ~50% of cases (↑ tyrosine kinase activity)
|
|
|
What is the prognosis of Primary Myelofibrosis (PMF)? Causes?
|
- Pre-fibrotic / early stage: > 10 years
- Fibrotic / typical stage: 3-7 years - BM failure (w/ infections and/or hemorrhage), thromboembolic events, portal HTN, or cardiac failure |
|
|
What can Primary Myelofibrosis (PMF) progress to? How common?
|
- 5-30% develop AML
- BM failure (w/ infections and/or hemorrhage), thromboembolic events, portal HTN, or cardiac failure |
|
|
Which Myeloproliferative Neoplasm is characterized by proliferation of Megakaryocytes?
|
Essential Thrombocytopenia (ET)
|
|
|
What are the peripheral blood abnormalities in Essential Thrombocytopenia?
|

- Elevated platelet count >450,000/µL
- Platelets have atypical morphology (large/giant, hypogranular) - Mild neutrophilic leukocytosis |
|
|
What are the Bone Marrow abnormalities in Essential Thrombocytopenia?
|
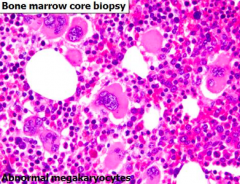
- Hypercellular
- Abnormal megakaryocytes |
|
|
What are the symptoms of Essential Thrombocytopenia?
|
- Bleeding tendency
- Thrombosis |
|
|
What genetic/molecular abnormality is associated with Essential Thrombocytopenia?
|
JAK2 mutation in 50% of cases (↑ tyrosine kinase activity)
|
|
|
What is the prognosis for Essential Thrombocytopenia?
|
Median survival: 12-15 years
|
|
|
How do you treat Essential Thrombocytopenia? Effect?
|
Alkylating agents or similar drugs to lower platelet count
|
|
|
What characterizes Myelodysplastic Syndromes (MDS)?
|
- Clonal hematopoietic stem cell disorder
- Cytopenias - Dysplasia (morphologic abnormalities) in one or more myeloid cell lineages - Ineffective hematopoiesis |
|
|
What myeloid cell lineages are affected by Myelodysplastic Syndromes (MDS)?
|
One or more myeloid cell lineages:
- Monocytes - Neutrophils - Eosinophils - Basophils |
|
|
What happens to the hematopoietic precursors in bone marrow in Myelodysplastic Syndromes (MDS)?
|
- Increased proliferation of hematopoietic precursors → hypercellular BM (but myeloblasts < 20%)
- Ineffective hematopoiesis and enhanced apoptosis → cytopenias in peripheral blood |
|
|
What are you at increased risk for if you have Myelodysplastic Syndrome (MDS)?
|
Acute Myelogenous Leukemia (AML) in 30% of cases
|
|
|
Which syndrome/disease is associated with cytoses? Cytopenias?
|
- Cytoses (↑ peripheral blood counts): Myeloproliferative Neoplasms
- Cytopenias (↓ peripheral blood counts): Myelodysplastic Syndromes |
|
|
What lab findings are associated with Myelodysplastic Syndromes (MDS)?
|
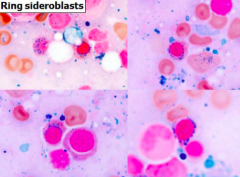
* Cytopenias (uni-, bi-, or pancytopenia)
- Leukoerythroblastic Reaction * Dysplastic features * Hypercellular BM - Ring sideroblasts - ↑ Myeloblasts - Chromosomal abnormalities in 50% |
|
|
What genetic abnormalities are associated with Myelodysplastic Syndromes (MDS)? How common?
|
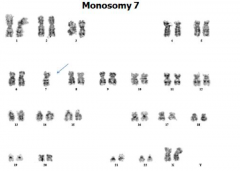
- Monosomy 5 or del5q
- Trisomy 8 - Monosomy 7 or del7q - 50% of cases |
|
|
What are Red Sideroblasts? When conditions are these associated with?
|
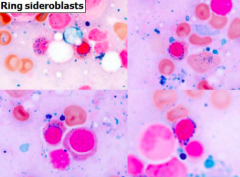
- Red cell precursors w/ frequent iron granules surrounding the nucleus
- Abnormal iron accumulation in functionally impaired mitochondria - Associated with Myelodysplastic Syndromes (MDS) and other non-neoplastic conditions like alcohol consumption, drugs (isoniazid), or other toxins |
|
|
Who is more likely to get Myelodysplastic Syndromes (MDS)?
|
Elderly (50-80 years)
|
|
|
What are the presenting symptoms of Myelodysplastic Syndromes (MDS)?
|
- Weakness
- Infections - Hemorrhage (d/t cytopenias) - OR asymptomatic |
|
|
What is the prognosis for Myelodysplastic Syndromes (MDS)? What can it progress to? Causes of death?
|
- Median survival: 9-29 months (t-MDS is 4-8 months)
- Progression to AML in 30% of cases - AML, infections, bleeding can all cause death |
|
|
How do you treat Myelodysplastic Syndromes (MDS)?
|
- Some get aggressive chemotherapy but it is difficult to cure and may be intolerable
Supportive therapy: - Blood products - Antibiotics - Growth factors * Hypomethylating agents Allogeneic stem cell transplant (potentially curing MDS) |
|
|
When are hypomethylating agents used? Effect?
|
Myelodysplastic Syndromes (MDS)
- They are not curative - Can reduce bone marrow cellularity, percentage of blasts, and improves cytopenias |
|
|
What is the effect of Allogenic Stem Cell Transplant in Myelodysplastic Syndromes (MDS)? Concerns?
|
- Only potential method to cure MDS
- Lack of matched donors and relatively high morbidity and mortality associated with this procedure limit those that may benefit from this |
|
|
What are Acute Leukemias? Chronic Leukemias?
|
- Acute: neoplastic proliferations of immature cells (blasts)
- Chronic: neoplastic proliferations of mature cells |
|
|
What are the types of Acute Leukemias?
|
- Myeloblastic or myeloid
- Lymphoblastic |
|
|
What is the natural history of untreated Acute Leukemias? Chronic Leukemias?
|
- Acute: weeks to months
- Chronic: months to years |
|
|
Which type of Acute Leukemia is more common in children? Adults? Males/Females?
|
- Children (<18y): 80-85% have ALL (can also occur in adults) (equal in males and females)
- Adults: 80-90% have AML (incidence increases with age, median age = 60 y) (slight male predominance) |
|
|
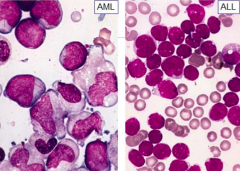
What clues do you make the differential diagnosis of AML vs ALL based on?
|
- Morphology
- Cytochemistry - Immunophenotyping - Genetics |
|
|
Acute Myelogenous Leukemia:
- Blast size - Chromatin - Nucleoli - Cytoplasm - Auer rods - Myelodysplasia |
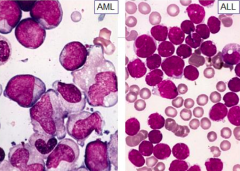
- Blast size: large and uniform
- Chromatin: finely dispersed - Nucleoli: 1 to 4 often prominent - Cytoplasm: moderately abundant, granules - Auer rods: 60-70% - Myelodysplasia: often |
|
|
Acute Lymphoblastic Leukemia:
- Blast size - Chromatin - Nucleoli - Cytoplasm - Auer rods - Myelodysplasia |
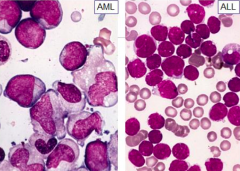
- Blast size: small/medium and variable
- Chromatin: coarse - Nucleoli: absent or 1 or 2; indistinct - Cytoplasm: scant to moderate, lacking granules - Auer rods: absent - Myelodysplasia: absent |
|
|
How does blast size compare for AML or ALL?
|
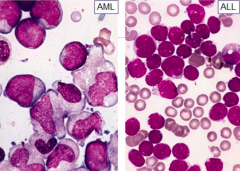
- AML: large and uniform blasts
- ALL: small/medium and variable blasts |
|
|
How do the chromatin compare for AML or ALL?
|
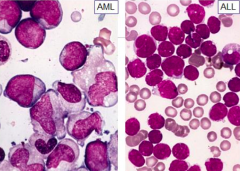
- AML: finely dispersed
- ALL: coarse |
|
|
How do the nucleoli compare for AML or ALL?
|
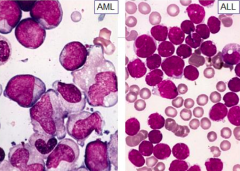
- AML: 1-4 often prominent
- ALL: absent or 1 or 2, indistinct |
|
|
How do the cytoplasm compare for AML or ALL?
|
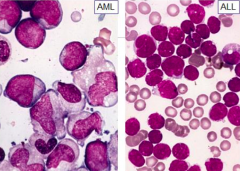
- AML: moderately abundant, granules often present
- ALL: scant to moderate, granules lacking |
|
|
How does the frequency of Auer rods compare for AML or ALL?
|
- AML: 60-70% of cases
- ALL: absent |
|
|
How does myelodysplasia compare for AML or ALL?
|
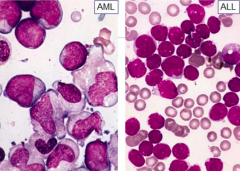
- AML: often present
- ALL: absent |
|
|
What does cytochemistry for diagnosing AML vs ALL rely on?
|
Takes advantage of presence of intracellular enzymes which generate a differently colored product when exposed to a substrate
|
|
|
What are the cytochemistry tests for diagnosing AML? What do they show if positive?
|

- Myeloperoxidase (MPO - generates a black intracellular product)
- Non-specific esterase (NSE - generates a brick-red intracellular product |
|
|
If you are concerned for AML and use a Myeloperoxidase (MPO) cytochemical test, what would be a positive result?
|
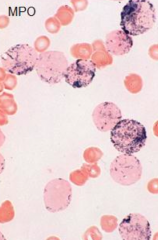
Generates a black intracellular product
|
|
|
If you are concerned for AML and use a Non-Specific Esterase (NSE) cytochemical test, what would be a positive result?
|
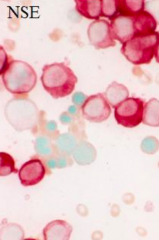
Generates a brick-red intracellular product
|
|
|
What are the methods of assessing for AML vs ALL with immunophenotyping?
|
- Immunohistochemistry
- Flow cytometry |
|
|
What are the benefits of immunophenotyping (immunohistochemistry or flow cytometry) in differentiating between AML and ALL?
|
- Differentiates between AML and ALL
- Identifies subtypes of AML - Used to establish a fingerprint for minimal residual disease assessment |
|
|
What is the method of Immunohistochemistry?
|
Uses tagged antibodies that recognize antigens expressed by cells in tissue sections
|
|
|
What is the method of flow cytometry?
|
- Uses tagged antibodies that recognize antigens expressed by cells in a cell suspension
- Assesses more than one antigen at at ime - Usually for antibodies designated by CD# |
|
|
How does immunohistochemistry (IHC) compare to Flow Cytometry?
|
Flow cytometry can assess more than one antigen at a time
|
|
|
What are the important antibodies to remember for AML? What are they markers of?
|
- CD34 and CD117
- Markers of immaturity |
|
|
What is the function of cytogenetics in assessing AML vs ALL?
|
Distinguishes sub-groups of AML, based on cytogenetic abnormalities that have biologic and prognostic significance
|
|
|
What is the prognosis for AML?
|
- Poor outcome
- Overall long-term survival of ~25% |
|
|
What kind of cells are involved in AML? Location?
|
- Myeloid progenitor cells
- Peripheral blood, bone marrow, may also involve extramedullary sites |
|
|
What are the symptoms of AML?
|
- Cytopenias cause weakness, fatigue, petechiae, infections
- Less common: organomegaly, lymphadenopathy, and infiltration of other extramedullary tissues - Coagulopathy in some variants (acute promyelocytic leukemia) |
|
|
Which subtype of AML has coagulopathy (clotting disorder)?
|
Acute Promyelocytic Leukemia
|
|
|
What is the main diagnostic criterion for AML?
|
Greater than 20% myeloid blasts in blood or marrow
|
|
|
What are the laboratory findings in AML?
|
- Myeloid blasts >20%
- Hypercellular BM - Severe leukopenia to marked leukocytosis - Anemia - Thrombocytopenia - Maturing cells of myeloid lineages may be dysplastic |
|
|
What is true of the blasts in AML?
|
- >20% in BM or PB
- Blasts have limited ability to mature beyond blast stage - Maturation may be towards any of the myeloid lineages (*granulocytes, *monocytes, megakaryocytes, or erythroid precursors) |
|
|
What are the types of AML?
|
- AML w/ recurrent cytogenetic abnormalities
- AML w/ myelodysplasia-related changes - Therapy-related myeloid neoplasms (including AML) - AML, not otherwise specified |
|
|
Why is it important to classify the types of AML?
|
Biological correlates to classification
|
|
|
What are the features of AML w/ recurrent cytogenetic abnormalities?
|
- Generally reciprocal translocations
- Generally flat incidence rate across age groups - Distinctive morphologic features - Dysplasia of maturing lineages is not prominent - No antecedent myelodysplastic syndrome FAVORABLE prognosis |
|
|
What are the features of AML w/ myelodysplasia-associated changes?
|
- Related biologically to MDS
- MDS type cytogenetic abnormalities (complex karyotypes, loss of chromosomes, or parts of chromosomes) - Increasing incidence w/ age - Prominent multilineage dysplasia POOR prognosis |
|
|
How does the prognosis compare for AML w/ recurrent cytogenetic abnormalities and AML w/ myelodysplasia-associated changes?
|
- AML w/ recurrent cytogenetic abnormalities: favorable prognosis
- AML w/ myelodysplasia-associated changes: poor prognosis |
|
|
What are the genetic abnormalities associated w/ AML w/ recurrent cytogenetic abnormalities?
|
- t(8;21)
- inv(16) - t(15;17) - PML/RARα gene fusion - 11q23 (MLL) rearrangement |
|
|
Which of the recurrent cytogenetic abnormalities associated w/ AML have a favorable prognosis?
|
- t(8;21)
- inv(16) - t(15;17) - PML/RARα gene fusion |
|
|
Which of the recurrent cytogenetic abnormalities associated w/ AML have an intermediate to unfavorable prognosis?
|
11q23 (MLL)
|
|
|
How common is AML w/ t(8;21)? Prominent features? Prognosis?
|
- 5-12%
- Prominent granulocytic maturation - Prominent Auer rods - Extramedullary involvement relatively common - Favorable prognosis |
|
|
How common is AML w/ inv(16)? Prominent features? Prognosis?
|
- 10-12%
- Similar to myelomonocytic leukemia - dual differentiation towards granulocytes and monocytes - BM contains eosinophils w/ dysplastic features - Extramedullary involvement is common - Favorable prognosis |
|
|
How common is AML w/ t(15;17)? Prominent features? Prognosis?
|
- 5-8%
- Acute Promyelocytic Leukemia (APL) - Atypical, hypergranular promyelocytes (rarely microgranular) - Reniform nuclei - Multiple prominent Auer rods ("****** cells") occassionally - Leukopenia assoc. w/ hypergranular/typical forms - Leukocytosis assoc. w/ microgranular forms - Disseminated intravascular coagulation (DIC) at diagnosis → early morbidity and martality - Favorable prognosis |
|
|
What fusion gene is generated by t(15;17)? How do you treat AML w/ t(15;17)?
|
- Generates the PML-RARα (retinoic acid receptor alpha) fusion gene
- Responds to all-trans Retinoic Acid (ATRA) - favorable prognosis - Matures the cells and also corrects the coagulopathy |
|
|
What are the implications of the formation of the PML-RARα fusion gene? What causes this?
|
- Caused by t(15;17) translocation
- Retinoic acid is important for myeloid maturation - Disrupting its receptor produces maturation arrest at promyelocyte stage - Treatment w/ all-trans Retinoic Acid (ATRA) overcomes the block and essentially matures the cells |
|
|
For what disease is ATRA (All-Trans Retinoic Acid) used?
|
t(15;17) variant of AML because this causes a PML-RARα fusion gene
|
|
|
How common is AML w/ 11q23 (MLL) rearrangements? Prominent features? Prognosis?
|
- 5-6% of AML
- Prevalent among infantile AMLs - Monocytic features - Hyperleukocytosis - Extramedullary tissue infiltration - t(9;11) translocation has intermediate prognosis - Other translocations involving MLL gene have intermediate to poor prognosis |
|
|
When does AML w/ myelodysplasia related changes occur? Prognosis?
|
- May follow an Myelodysplastic Syndrome (MDS)
- May occur without antecedent MDS - Primarily in older adults - Unfavorable prognosis |
|
|
What are the prominent features of AML w/ myelodysplasia related changes?
|
- Dysplasia in >50% of 2+ lineages AND/OR
- Myelodysplasia-associated cytogenetic abnormalities (eg, monosomy 7 / del(7q), monosomy 5 / del(5q), or complex karyotypes) - Unfavorable prognosis |
|
|
What are the myelodysplasia associated cytogenetic abnormalities? Prognosis?
|
- Monosomy 7 / del(7q)
- Monosomy 5 / del(5q) - Complex karyotypes - Unfavorable prognosis |
|
|
What can cause therapy-related AML and MDS?
|
- Patients previously treated w/ chemo and/or radiotherapy
- Two main classes of chemo drugs: alkylating agents and topoisomerase II inhibitors |
|
|
What can alkylating agent and topisomerase II inhibitor chemotherapies both lead to an increased incidence of?
|
Therapy-Related AML and MDS
|
|
|
When does Alkylating Agent Related AML/MDS occur? What is risk for developing this related to?
|
- Occurs ~5 years after treatment
- Risk related to total dose and patient age |
|
|
What are the features and prognosis of Alkylating Agent Related AML/MDS?
|
- Multi-lineage dysplasia
- Cytogenetic abnormalities (aberrancies of chromosomes 5 and 7) in nearly 100% of cases - Very poor prognosis - Similar to AML w/ myelodysplasia-related changes |
|
|
What type of AML is Alkylating Agent Related AML/MDS similar to?
What type of AML is Topisomerase II Inhibitor Related AML/MDS similar to? |
AML w/ myelodysplasia related changes
De Novo AML w/ 11q23 (MLL) gene rearrangements |
|
|
When does Topoisomerase II Inhibitor Related AML/MDS occur?
|
Occurs 2.5-3 years after exposure
|
|
|
What are the features and prognosis of Topoisomerase II Inhibitor Related AML/MDS?
|
- Monocytic or myelomonocytic morphology
- 11q23 (MLL) balanced translocations are most common cytogenetic abnormalities - Poor prognosis - Similar to De Novo AML w/ 11q23 (MLL) gene rearrangements |
|
|
How do you categorize AML if it does not fulfill criteria of the other three groups (AML w/ recurrent cytogenetic abnormalities; AML w/ myelodysplasia-related changes; or therapy-related AML)?
|
AML not otherwise specified (NOS)
|
|
|
How do you categorize AML not otherwise specified (NOS)?
|
- 3 categories based on degree of maturation
- 5 categories based on lineage of differentiation |
|
|
What kind of tumor is a Myeloid Sarcoma?
|
Extramedullary tumor of immature myeloid cells
|
|
|
How does Myeloid Sarcoma related to AML?
|
Myeloid Sarcoma can occur after, with, or preceding an AML diagnosis
|
|
|
How do you treat Myeloid Sarcoma? Why?
|
- Treat in same fashion as AML
- Because it is assumed that progression to systemic disease would eventually occur even if BM and blood are sometimes not overtly involved |
|
|
What are the variety of responses to treatment for AML?
|
- Ranges from cure to refractory disease
- Prognosis in individual patients cannot be estimated accurately, meaning that very good patients may relapse while very poor patients may be cured |
|
|
What are the new prognostic markers for AML that are attempting to refine the risk stratification? Prognosis?
|
- FLT3 - poor prognosis
- NPM1 - favorable prognosis |
|
|
What kind of molecule is FLT3? Marker of what?
|
- Receptor Tyrosine Kinase
- Has a role in hematopoietic progenitor survival and proliferation - Activating mutation caused by internal tandem duplications of juxtamembrane domain occurs in ~1/3 of AMLs → POOR prognosis - New prognostic marker for AML (seen in various subtypes) |
|
|
What kind of molecule is NPM1? Marker of what?
|
- Nucleocytoplasmic shuffling protein
- Mutated NPM1 seen in 50-60% of AML w/ NORMAL karyotype - Favorable prognosis in cases w/o mutated FLT3 |
|
|
What happens if you have both FLT3 and NPM1 mutations?
|
Mutated FLT3 (poor prognosis) trumps the good prognostic effect of a mutated NPM1
|
|
|
What do "histiocytoses" refer to?
|
Variety of proliferative disorders of dendritic cells or macrophages
|
|
|
How severe are Histiocytoses?
|
- Some are very rare and highly malignant
- Others are completely benign and reactive |
|
|
What are the types of Histiocytoses?
|
- Histiocytic Lymphomas (very rare and malignant)
- Histiocytic proliferations in lymph nodes (benign and reactive) - Langerhans Cell Histiocytoses (tumors made of Langerhans cells) |
|
|
What are Langerhans cells? Where? Function?
|
- Immature dendritic cells found in the epidermis
- Function to capture antigens and display them to T cells |
|
|
What do Langerhans cells express in Langerhans Cell Histiocytoses?
|
- MHC Class II antigens
- CD1a - Langerin |
|
|
What is Langerin? Where is it found? What does it look like?
|
- Transmembrane protein
- Found in Birbeck granules (cytoplasmic pentalaminar rodlike tubular structures) - Look like tennis rackets (characteristic periodicity and a dilated terminal) |
|
|
What do proliferating Langerhans cells in Langerhans Cell Histiocytoses look like?
|
- Do not resemble their normal dendritic counterparts
- Instead, they have abundant, often vacuolated cytoplasm and vesicular nuclei - Appearance more akin to tissue macrophages (called histiocytes) |
|
|
What are the three relatively distinct clinicopathology entities of Langerhans Cell Histiocytoses?
|
- Multi-system Langerhans Cell Histiocytoses (Letterer-Siwe Disease)
- Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma) |
|
|
When is Multi-system Langerhans Cell Histiocytoses (Letterer-Siwe Disease) more likely to occur? Signs?
|
- Children < 2y
- Multifocal cutaneous lesions that grossly resemble seborrheic skin eruptions - Hepatosplenomegaly - Lymphadenopathy - Pulmonary lesions - Later, destructive osteolytic bone lesions |
|
|
What happens to the skin in Multi-system Langerhans Cell Histiocytoses (Letterer-Siwe Disease)?
|
- Multifocal cutaneous lesions
- Grossly resemble seborrheic skin eruptions - Composed of Langerhans cells |
|
|
What happens to the bone marrow in Multi-system Langerhans Cell Histiocytoses (Letterer-Siwe Disease)? Implications?
|
- Extensive infiltration
- Leads to pancytopenia - Predisoposes to recurrent infections like otitis media and mastoiditis |
|
|
What is the prognosis of Multi-system Langerhans Cell Histiocytoses (Letterer-Siwe Disease)? Treatment?
|
- Rapidly fatal if untreated
- With extensive chemo, 50% survive 5 years |
|
|
What are the characteristics of Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma)? Location of lesions?
|
- Unifocal (skeletal system) or multifocal
- Expanding, erosive accumulations of Langerhans cells, usually within medullary cavities of bones (or less commonly in skin, lungs, or stomach) - Any bones in skeletal system: calvaria, ribs, femur most commonly |
|
|
What do Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma) lesions contain?
|
Langerhans cells, admixed w/ variable numbers of lymphocytes, plasma cells, neutrophils, and eosinophils, which are usually, but not always prominent
|
|
|
What are the symptoms of UNIFOCAL Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma)? Treatment?
|
- Most often involves the skeletal system
- Asymptomatic or may cause pain, tenderness, and pathologic fractures - Indolent disorder that may heal spontaneously or be cured by local excision or irradiation |
|
|
What are the symptoms of MULTIFOCAL Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma)? Treatment?
|
- Usually affects children
- Typically manifests w/ multiple erosive bony masses that sometimes extend into the soft tissues - 50% involve posterior pituitary stalk of hypothalamus which leads to Diabetes Insipidus - Calvarial bone defects + Diabetes Insipidus + Exophthalmos = Hand-Schüller-Christian Triad - Many have spontaneous regressions, others treated effectively w/ chemotherapy |
|
|
What is the Hand-Schüller-Christian Triad? When is it seen?
|
Combination of:
- Calvarial bone defects - Diabetes Insipidus - Exophthalmos (abnormal protrusion of the eyeball or eyeballs) Associated with Multifocal Uni-system Langerhans Cell Histiocytoses (Eosinophilic Granuloma) |
|
|
What genetic abnormality is associated with the Langerhans cell tumors?
|
- Acquired mutation in serine/threonine kinase BRAF
- Valine to glutamate substitution in residue 600 that leads to hyperactivity of kinase - BRAF is a component of Ras signaling pathway that drives cellular proliferation and survival, effects that likely contribute to the growth of the noplastic Langerhans cells |
|
|
What is an acquired mutation in serine/threonine kinase BRAF associated with?
|
- Langerhans cell tumors (Histiocytoses)
- Hairy cell leukemia - Benign nevi - Malignant melanoma - Papillary thyroid carcinoma - Some colon cancers |
|
|
What kind of condition is Hemophagocytic Lymphohistiocytosis (HLH)?
|
Potentially fatal hyper-inflammatory condition
|
|
|
What can cause Hemophagocytic Lymphohistiocytosis (HLH)?
|
- May occur as primary (genetic) condition d/t mutations in genes (Eg, perforin) important in the cytolytic secretory pathway
- Causes perforin and granzymes to induce apoptosis in target cells * Familial HLH (HLH is the only manifestation of the disease) * Other genetically caused HLH (HLH is one of several clinical manifestations) |
|
|
Identical clinical findings of Hemophagocytic Lymphohistiocytosis (HLH) may be caused by what?
|
Secondary to infectious, rheumatologic, malignant, or metabolic conditions = Secondary HLH
|
|
|
How does the type of Hemophagocytic Lymphohistiocytosis (HLH) affect treatment?
|
Whether primary (familial) or secondary (infectious, rheumatologic, malignant, or metabolic), HLH therapy needs to be instituted promptly to prevent irreversible tissue damage
|
|
|
What end of the spectrum is Hemophagocytic Lymphohistiocytosis (HLH) of the hyper-inflammatory disorders?
|
Severe end of spectrum when the immune system starts to damage host tissues
|
|
|
What are the clinical features diagnostic of Hemophagocytic Lymphohistiocytosis (HLH)?
|
There is no single clinical feature alone diagnostic of HLH, you need the entire clinical presentation to make the diagnosis
|
|
|
What is hemophagocytosis? How does this relate to Hemophagocytic Lymphohistiocytosis (HLH)?
|
- Hallmark of activated macrophages
- Neither specific nor sensitive for HLH, its presence should only be considered supportive of HLH |
|
|
What can hemophagocytosis be indicative of?
|
- Supportive of a diagnosis of Hemophagocytic Lymphohistiocytosis (HLH)
- Also seen as secondary disorder in association with severe infections, malignancies, rheumatologic disorders, and some metabolic diseases |
|
|
What is the most common infectious cause of Hemophagocytic Lymphohistiocytosis (HLH)?
|
EBV - may trigger HLH in patients w/ familial disease
|
|
|
What is the most common malignant cause of Hemophagocytic Lymphohistiocytosis (HLH)?
|
Lymphoma (most common) or leukemia of T or NK cell lineages
|
|
|
What is the prognosis for Familial Hemophagocytic Lymphohistiocytosis (HLH)?
|
- Before the initiation of modern therapeutic regimens, 1-year survival rate of children was close to 0%
- With introduction of stem cell transplant (SCT) and increased experience, survival has improved to 92% (delay in diagnosis and multi-organ involvement associated w/ worse prognosis) |
|
|
What is the prognosis for the different types of Secondary Hemophagocytic Lymphohistiocytosis (HLH)?
|
Mortality varies from:
- 8-22% in rheumatologic-HLH (better prognosis) - 18-24% in EBV-HLH (worse prognosis) (delay in diagnosis and multi-organ involvement associated w/ worse prognosis) |
|
|
Which condition is associated with a very high Ferritin?
|
Hemophagocytic Lymphohistiocytosis (HLH)
|