Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
15 Cards in this Set
- Front
- Back
|
Diagnostic criteria: (a) 100+ colorectal polyps, (b) germ line mutation in APC gene, (c) family history of APC and (d) at least one of epidermoid cyst, osteoma or desmoid tumor
|
Familial adenomatous polyposis of colon-classic
|
|
|
Colonic adenomatous polyps plus extraintestinal features of multiple osteomas (skull, mandible, long bones), epidermal cysts, fibromatosis
|
Gardner’s syndrome and colon
(variant of familial adenomatous polyposis) |
|
|
Diagnostic criteria: (a) 6 or more colorectal juvenile polyps, (b) juvenile polyps throughout entire GI tract, or (c) any number of polyps in a patient with a family history of juvenile polyposis
|
Juvenile polyposis syndrome and colon
|
|
|
often mutations in PTEN, SMAD4/DPC4/MADH4
|
Juvenile polyposis syndrome and colon
|
|
|
Rare autosomal dominant disorder with < 100 adenomas, usually in proximal colon
Multiple colorectal tumors associated with multiple sebaceous tumors, keratoacanthomas and basal cell carcinoma 15% of women develop endometrial cancer |
Muir-Torre syndrome and colon
|
|
|
Diagnostic criteria: (a) 3+ histologically confirmed Peutz-Jeghers polyps, (b) any Peutz-Jeghers polyp with a family history, (c) characteristic melanotic mucocutaneous pigmentation (lips, oral mucosa, genitalia, digits, palms, soles) with a family history or (d) Peutz-Jegher polyp and characteristic mucocutaneous pigmentation
Hamartomatous polyps are present throughout GI tract excluding esophagus |
Peutz-Jeghers syndrome and colon
|
|
|
Rare; autosomal recessive variant of familial adenomatous polyposis
CNS tumors (gliomas or glioblastomas) and later familial adenomatous polyposis May be 2 types - (a) gliomas plus nonpolyposis adenomas with mismatch repair gene mutations and (b) medulloblastomas plus adenomatous polyposis with APC mutations |
Turcot’s syndrome and colon
Pronounced with silent second t (Turcot was French Canadian) Rare; autosomal recessive variant of familial adenomatous polyposis |
|
|
Harmatomas/neoplasms of face, thyroid, and GI tract
Autosomal dominant (with incomplete penetrance and variable expressivity) with facial tricholemmomas, acral keratoses, oral mucosal papillomas and colorectal polyps |
Cowden's syndrome and colon
|
|
|
Usually age 50-60 yearsRare, non-hereditary disorder of multiple GI juvenile type polyps, usually colonic, associated with ectodermal changes (alopecia, nail atrophy, hyperpigmentation), protein losing enteropathy with associated diarrhea
|
Cronkhite-Canada syndrome and colon
|
|
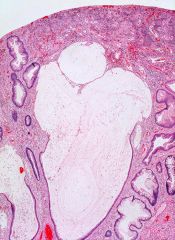
|
Juvenile polyp
Irregularly shaped tubules and cysts lined by bland Kolonepithel are embedded in abundant stroma. The surface is eroded and the stroma is replaced by granulation tissue. |
|
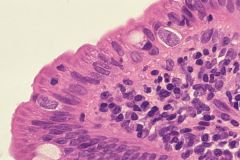
Duodenal biopsy from a patient suffering from a long-standing intractable diarrhea
|
Isospora belli infection
|
|

Duodenal biopsy from a patient suffering from a long-standing intractable diarrhea
|
Perforation at the site of Peyer patches is a serious complication of typhoid fever
|
|
|
Which of the following cystic lesions of the liver communicate with the intrahepatic biliary tree?
a. Solitary (non-parasitic) cyst b. Hydatid (echinococcal) cysts c. Polycystic liver disease d. Caroli disease e. Cyst caused by Entamoeba hystolytica |
d.
Hepatobiliary cysts can be parasitic, developmental, or undetermined origin. The most common type is solitary non-parasitic simple cyst. Cysts in Caroli disease communicate with the rest of the biliary tree. In other cystic lesions on the list, cysts do not communicate with the biliary tree in general. Ascending cholangitis can result from the communications between cysts and the biliary tract system. Caroli disease (congenital dilatation of the intrahepatic bile ducts) generally involves the entire liver. Microscopically, severe chronic inflammation, with or without superimposed acute inflammation, luminal inspissated mucin and bile are common findings in this disease. |
|
|
The most common form of congenital extrahepatic bile duct dilatation is:
a. Caroli disease b. Choledochal cyst c. Choledochocele d. Diverticulum of the common bile duct e. Cholangiectases |
b. Choledochal cyst
is a congenital cystic dilatation of the extrahepatic biliary tree, intrahepatic biliary radicles, or both and commonly presents with the triad of pain, jaundice, and mass in the right upper quadrant. They are more prevalent in Asia than in the United States and other Western countries. Caroli disease is characterized by dilatation of the intrahepatic biliary tree. Caroli disease is sporadic, whereas Caroli syndrome is generally inherited in an autosomal recessive manner. The latter is often associated with congenital hepatic fibrosis and portal hypertension. This form of Caroli disease is also often associated with autosomal recessive polycystic kidney disease (ARPKD). Diverticulum of the common bile duct and choledochocele are less common than choledochal cyst. Cholangiectases are not congenital and usually are caused by inflammatory process. |
|

|
Epithelioid hemangioendothelioma. Proliferation of vascular channels lined by atypical epithelioid cells. Notice the sclerotic background.
|