![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
24 Cards in this Set
- Front
- Back
|
The defects of which TCA cycle enzymes were discussed during the last demonstration lab?Why these? |
- Pyruvate dehydrogenase complex (PARTIAL defect) - Pyruvate carboxylase deficiency (A,B,C-types) - Defects in other TCA enzymes results in death (most often at fetus stage) |
|
|
Write with structures the reaction that couples glycolysis and TCA cycle. List the cofactorsalso. |
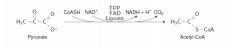
Pyruvate dehydrogenase complex |
|
|
Name the four glycogenoses occurring in the material of the previous demo lab. |
- Type 1: Von Gierke´s disease - Type 2: Pompe´s disease - Type 3: Cori´s disease - Type 5: McArdle´s disease |
|
|
Name the two different activities of the debranching enzyme. Which of these is defective inCori disease? |
- Glucosidase activity - Glucosyltransferase activity - Cori´s disease: Glucosidase activity is defective! |
|
|
Under what conditions and why is the partial defect of glucose 6-phosphate dehydrogenasean evolutionary advantage? |
In the case of malaria. There is higher oxidative stress in the RBC (due to less GSH regeneration) which proves fatal to plasmodium. |
|
|
Why does glucose-6-phosphate dehydrogenase deficiency cause serious problem for the redblood cell? |
- Regeneration of GSH from GSSG requires NADPH - The pentose phosphate pathway (G6PD is the rate-limiting step) is the only source of reduced Glutathione (GSH) in RBC. The lack of GSH puts the RBC at a substantial risk of damage from reactive oxygen species, due RBC being oxygen carriers and lacking the ROS-protective functions of reduced Glutathione (antioxidant) -> hemolytic anemia |
|
|
The defect of an enzyme causes the accumulation of homogentisate. Write with structuresthe reaction and name the related disease. |
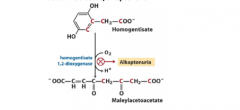
Black urine disease/alkaptonuria |
|
|
List three lysosomal storage diseases. |
- Tay-Sachs disease - I-cell disease - Niemann-Pick disease |
|
|
What enzymes are necessary for the degradation of fructose in human liver? |
- Fructokinase - Fructose-1-phosphate aldolase - Triose kinase - (Triose phosphate isomerase) |
|
|
Name 5 metabolic diseases with a characteristic symptom of metabolic acidosis. |
- Methylmalonic acidemia - Propionic acidemia - Ketoacidosis - Lactic acidosis - Diabetic acidosis |
|
|
Write with structures the reaction defective in fructosuria. |
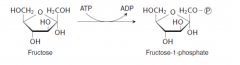
Hepatic fructokinase |
|
|
Write with structures the reaction defective in fructosaemia. |
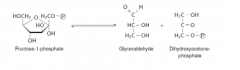
Fructose-1-phosphate aldolase |
|
|
Write with structures the reaction defective in alkaptonuria. |
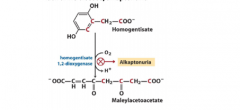
Black urine disease/alkaptonuria |
|
|
Write with structures the reaction defective in von Gierke’ disease. |
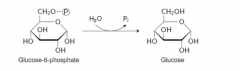
Glucose-6-phosphatase |
|
|
Write with structures the reaction defective in McArdle’s disease. |

|
|
|
Name the enzyme defective in albinism and name the required cofactor as well. |
- Tyrosinase - Cu2+ |
|
|
Name the two compounds which are directly bound to form (gluco)cerebrosids. |
- Glucose - Ceramide |
|
|
Name the common precursor of cerebroside synthesis and glycogen synthesis. |
UDP-glucose |
|
|
Name the starting compounds of the (gluco)cerebrosid synthesis. |
- Palmitoyl-CoA - L-serine |
|
|
Name the two compounds which are directly bound to form sphingomyelins. |
- Ceramide - Phosphocholine/(Phosphoethanolamine) head group |
|
|
Name the starting compounds of the sphingomyelin synthesis. |
- Palmitoyl-CoA - L-serine |
|
|
Name the two compounds which are bound to form gangliosides. |
- Glucosphingolipid - Sialic acid |
|
|
List the possible causes of familiar hypercholesterolaemia. |
- Missing/non-functional LDL-receptors - Mutation in ApoB100 - Instead of liver cells macrophages take up cholesterol -> foam cells |
|
|
List the lipoproteins. Indicate the one containing the most cholesterol. |
- LDL contains the most cholesterol - VLDL - IDL - HDL - Chylomicrons (contain the most triglycerides) |