![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
64 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
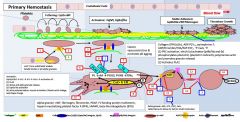
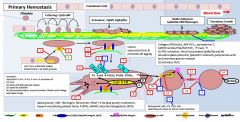
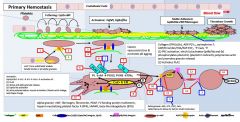
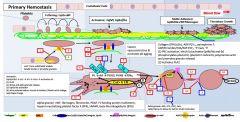
Primary vs secondary hemostasis |
Primary: platelets/fibrin/clot Secondary: coagulation factors |
|
|
|
Match symptom with primary vs secondary hemostatic defect: A. petechiae B. small ecchymoses C. hemarthosis D. menorrhagia E. deep soft tissue hematomas F. extensive postop bleed G. delayed onset of bleeding |
Primary: menorrhagia, petechiae, bleeding immediately follows trauma, spreading deep soft tissue hematomas [A, D] Secondary: hemarthrosis, deep soft tissue ecchymoses, delayed/extensive post-op bleed, gingival bleeds [B, C, E, F, G] |
|
|
|
Symptoms of a primary hemostatic disorder |
Mucosal (gingival/epistaxis) and cutaneous bleeding (petechiae,ecchymoses) Menorrhagia/metrorrhagia Patients tend to bleed immediately after vascular trauma |
Ecchymoses: usually develop without noticeable trauma and DO NOT spread into deeper tissues Menorrhagia/metrorrhagia: 15%caused by bleeding diathesis (vWD, ITP, platelet function defect) |
|
|
A patient has petechiae with normal platelet counts and PT/PTT. What's the next step? |
PFA-100 |
|
|
|
What do the following have in common? Factor V Leiden Protein S deficiency Protein C deficiency AntithrombinIII deficiency Prothrombin mutation (G20210A) Lupus anticoagulant Pregnancy Plasmin defects |
Hypercoagulable states/diseases |
|
|
|
Wilms tumor and coagulation |
Associated with acquired vWD |
|
|
|
Thrombomodulin plus F2a = ? |
Activates protein C PC --> APC |
|
|
|
Factor V Leiden results in what symptoms? |
Hypercoagulable state |
|
|
|
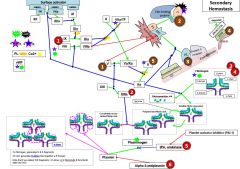
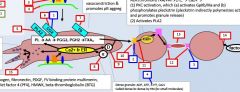
In primary hemostasis, platelets tether to areas of injury via __ (platelet) connections to __. |
Gp1b binds vWF |
|
|
|
Where does vWF bind? |
Binds collagen and Gp2b/3a |
|
|
|
Where does vWF come from? |
Platelet alpha granules and endothelial Wiebel Palade bodies |
|
|
|
What is GpIV? |
A collagen receptor |
|
|
|
Together with GpIV, GpIb leads to intracellular release of _. |
Calcium |
|
|
|
What does calcium do? |
Activates PLA2 (PL --> AA) Alpha and dense granule release PKC activation - activates Gp2b/3a) - actin polymerization |
|
|
|
PKC activation leads to __. |
Activates Gp2b/3a) Promotes actin polymerization |
|
|
|
PLA2 is activated by _. |
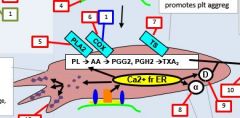
Calcium |
|
|
|
PLA2 takes _ and forms _. |
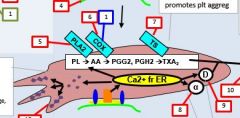
PL AA |
|
|
|
AA forms into __ by the action of __. |
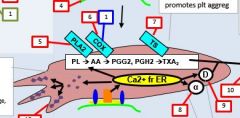
Prostaglandin COX |
|
|
|
Alpha granules contain _. |
vWF, fibrinogen |
|
|
|
Dense granules contain _. |
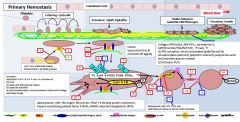
5HT, ADP, Ca2+ |
|
|
Name the disease associations (1-15) |
1) vWD 2) Bernard Soulier 4) Ehlers Danlos, Marfans 7) Thromboxane synthase def 8) Alpha storage pool disorders (gray plt syndrome) 9) Delta storage pool disease 12) Glanzmann thrombasthenia 13) Congenital afibrinogenemia 14) Scott syndrome: defect in flippase 15) Wiskott-Aldrich syndrome |
3) Collagen R defects 5) Impaired liberation of AA 6) COX def 10) Purine /TXA2receptor or G protein defects 11) Defects in PIP metabolism |
|
Glanzmanns |
12 |
|
|
Bernard Soulier |
2 Gp1b is 2nd most expressed integrin (25K on plt surface, MC is GpIIb/IIIa at 50K) |
|
|
Scott syndrome |
14) Scott syndrome: defect in flippase,leaves PS on inner leaflet, phosphatidyl choline on outside --> factors can’t kick start coagulation cascade |
|
|
Wiskott-Aldrich syndrome |
15) Wiskott-Aldrich syndrome- a defect in cdc42 geneblocks GpIb-mediatedfilopodiaformation (Gp1b --> cdc4(g protein) --> WASP --> actin polymerization). It also hassomething to do w granule release. This dz also affects T cells. |
|
|
dxby EM, difficult to test for bc hard to keep NL plts fr degranulating ImKuzBvanMcQ7cooki@ |
8)Alpha storage pool disorders (gray platelet syndrome) |
|
|
eptifibatide |
3) abciximab, tirofiban, eptifibatide-inhibit fibrinogen binding to GpIIb/IIIa, agonists in the PFA will not induceaggregation (weo ristocetin),these drugs are far more potent than aspirin, ITP (2/2 to drug-dependent Abs)is a possible sdeeffect |
|
|
prasugrel |
2) clopidogrel, ticlodipine,prasugrel- thienopyridinederivatives that inhibit the ADP P2Y12 receptor, they also affect plt response to collagen, epinephrine, and thrombin |
|
|
ibuprofen |
1) Aspirin (irreversible- takes 5-7d fordrug to wear off- the lifespan of plts), NSAIDs (reversible- ibuprofen, forexample, only takes 24h to wear off). Note: though aspirin irreversible, itonly blocks one of many pathways, thus, plts are NOT totally inactivated |
|
|
SSRI |
4) SSRIs: pts may be more prone to GIT or post-op bleed |
|
|
clopidogrel |
2 |
|
|
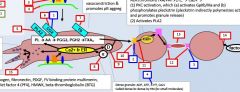
tirofiban |
3 |
|
|
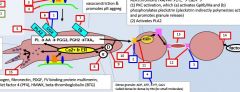
ticlodipine |
2 |
|
|
|
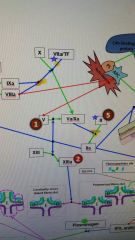
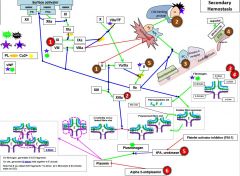
F12 is activated by _. |
F2a |
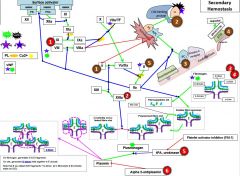
|
|
|
12a activates _. |
11 |
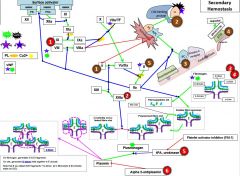
|
|
|
11a activates _. |
9 |
|
|
|
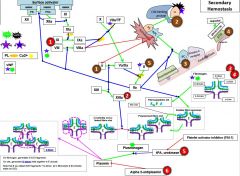
8 is activated by _. |
2 |
|
|
|
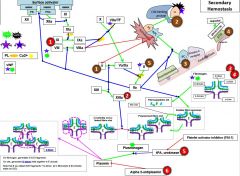
10 is activated by _. |
VIIa/TF 9a/8a |
|
|
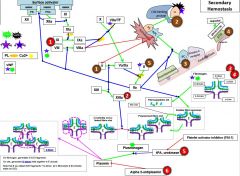
Red 1 |
Hemophilia |
|
|
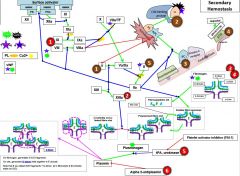
Red 2 |
Factor 13 deficiency |
It is diagnosed by urea clot solubility-non-covalent fibrin clot is soluble in urea, FXIII defproven when clot solubilizes to 5M urea |
|
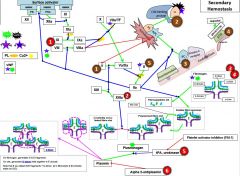
Red 3 |
3a) Hypofibrinogenemia-heterozygote, low but not usu clin signiffibrinogen. 3b) afibrinogenemia arehomozygous or compound heterozygous for fibrinogen alpha chain gene mutations.PT/PTT generally not affected bc fibrinogen is not a rate limiter. 4) Dysfibrinoginemia-fibrinogen polymerizes slowly, activity usu ↓/NL but [fibrinogen] is ↑/NL when measured immunologically (eg,radial immunodiffusion,ELISA, or nephelometry). Dx: lowratio of functional to immunologic fibrinogen. 60% will not have sx butPT/PTT may be funny, 25% bleed, 10% thrombose(fibrin polymerizes & breaks-down slowly), 5% show pregcomplications e |
|
|
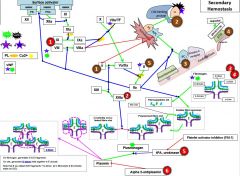
Red 5 |
5) In prostate cancer, incr urokinaseleads to bleeding. This is a rare example of bleeding in cancer, as cancer usu a.w hypercoagulablestates |
|
|
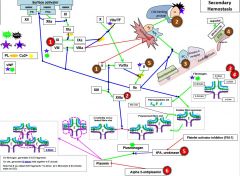
Red 6 |
6) Alpha 2-antiplasmin(A2AP) deficiency- can be congenital or acquired (cirrhosis, also seentransiently following liver transplant as it takes time for liver fxn tocome back) |
|
|
|
Hypercoagulable states lead to _ thrombi. A. venous B. arterial C. both |
A. Venous Note: Arterial TE are not usu associated with hypercoagstates- these are caused by atherosclerosis/trauma |
|
|
|
Prothrombin (G20210A) mutation causes _. |
Hypercoagulable state |
|
|
|
Protein S is bound in serum to _. |
C4b binding protein |
|
|
|
Protein S does what? |
S for Support It support F5/8 cleavage by activated protein C (APC) |
|
|
|
Protein C is activated by _. |
Thrombomodulin |
|
|
|
5M urea does what? |
Causes polymerized fibrin clots to become solube. Covalent linked fibrin does not so it can be used to test F13 deficiency. |
|
|
|
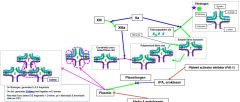
How do F2 and F13 work regarding fibrin clot formation? |
F2a snips fibrinopeptides from fibrinogen to produce D to E linkage F13a creates covalent D to D links |
|
|
|
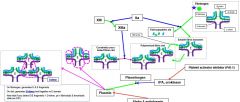
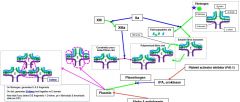
Plasminogen is activated by _. It is inhibited by _. |
tPA, urokinase Platelet activator inhibitor (PAI-1) Alpha 2-antiplasmin |
|
|
|
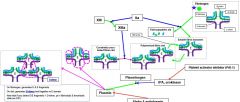
Plasmin cleaves _ into _. |
Onfibrinogen, generates D & E fragmentsOnclot, generates D-dimerheld together w E domainNotethat if you detect D/E fragments + D-dimer, pt in fibrinolytic & thrombotic state (ex DIC) |
|
|
|
Aminocaproic acid |
antifibrinolytic,used to tx bleeding Tranexamicacid-same MOA, 8Xeffective |

|
|

Warfarin |

Blue 1 |
|
|

Blue 2? |
2) Heparin: UFH: binds AT3 to 2a/9a/10a/11a/12a (UFH-T1/2 1.5h)LMWH: binds Xa alone (lovenox/enoxaparin(LMWH, T1/2 4-5h), fondaparinux(T1/2 17-21h)) |
|
|
|
Protamine sulfate can neutralize _. |
Protamine sulfate can neutralize UFH/LMWH but NOT fonda |
|
|
|
HIT |
HIT most likely to occur w UFH, some w LMWH, not at all w fondaparinux - |
|
|

lepirudin |
4) Direct thombin inhibitors: lepirudin(irreversible, T1/2 90-120 min, renal metab, in CKD T1/2 52h!), bivalirudin(reversible), agatroban(short-acting, reversible, liver metab-good for pts w CKD), dabigitran, all do not cause HITa(TM)"R! |
|
|

rivaroxaban |
3) Direct Xa inhibitors: rivaroxaban, apixaban(both have xa inname), irreversible, renal metab, T1/2 15h, metabd byCYP3A4 |
|
|
|
Mixing study |
•combinedpt plasma w plasma known to have 100% activity of all factors in 1:1 or 4:1ratio. –IfPT/PTT normalize, a factordeficiencyis at play. If it doesn’t, an inhibitor ispresent.–Note:if PT is prolonged, a mixing study is rarely performed bc an inhibitor to FVIIis so rare–4:1pt to ctrl can be useful for when weak inhibitors are present, as these maycorrect with a 1:1 mix. |
|
|
|
Euglobulin lysis time (ELT) |
An old test, rarely usedAcidify pt plasma à precipitate forms that is devoid ofATIII, alpha 1-antiplasmin, alpha 1-antitrypsinExtract precipitate and re-dissolveAdd thrombin in 37C water bath for 2 hrIf clot falls apart, it means there is aproblem in clot stabilityIt cannot give you an exact diagnosisSome patients will be normal foreverything except this test, hard to say what to do... |
|
|
|
Anti-phospholipid syndrome clinical criteria |
Clinicalcriteria:1 ormore premature (<34w) 2/2eclampsia-preE orplacental insufficiency1 ormore preglosses >10 wksEGA**3 ormore preglosses <10 |
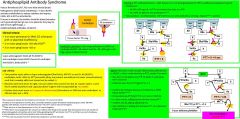
|
|
|
Anti-phospholipid syndrome lab criteria |
Twopositive tests, either a lupus anticoagulant (StaClotLA, dRVVT) or anti-PL Ab (B2GP1, cardiolipin, both>40IU or 99th percentile (they are present normallybut in lower concentrations), such that a weakly abNL test should not be called +)Mustbe same test (even for IgM, not sure where this comes from but Dr. Gailani saidit, I can’t find it stated anywhere and Laposata doesn’t agree with it) separated by >12weeksPositivetests must occur w.i 5years of clinical event (thrombosisin 2000 but anti-cardiolipin+ in 2014x2 does not = APS):#1 |
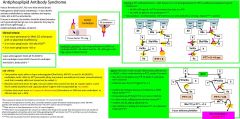
|
|
|
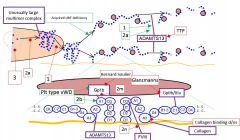
vWD factors to consider |
vWF antigen Ristocetin Cofactor CoF to antigen ratio F8 levels Multimer analysis RIPA Platelet count |

|