Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
543 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
General Carbohydrate Metabolism
|
CHO-->Glucose-->Glycolysis-->Acetyl CoA-->TCA-->Oxidative Phosphorylation
|
|
|
|
General Fatty Acid Metabolism
|
FAs-->Acetyl CoA-->TCA
-->Oxidative Phosphorylation |
|
|
|
General Protein Metabolism
|
Proteins-->AAs-->TCA--> Oxidative Phosphorylation
|
|
|
|
Glycolysis
|
Occurs in Oxygen depletion. Creates less ATP than OXPHOS
|
|
|
|
FA Usage
|
Not used by brain. First priority for Muscle. If FFA are very available to muscle, glycolysis decreases.
|
|
|
|
Keto Acids Usage
|
First priority for brain. Not used by muscle unless FA unavailable
|
|
|
|
Glucose Usage
|
Used most of the time (due to Keto Acid scarcity) in Brain. Used in muscle only if FA and Keto acids are unavailable.
|
|
|
|
Fuel Storage
|
Stores(days/months)
Meals (hours) Blood(minutes) ATP(seconds) |
|
|
|
Fuel storage CHO
|
CHO-->Glucose-->Glycogen (liver and muscles)
|
|
|
|
Fuel Storage Proteins
|
Proteins-->AAs-->muscle (and other proteins)
|
|
|
|
Fats Storage
|
Fats-->FAs-->Triglycerides--> Adipose tissue
|
|
|
|
CHO as Energy Source
|
Largest portion of dietary calories but one mole glucose yields only 34-36 ATP
|
|
|
|
Fats as energy source
|
Most efficient fuel. One mole palmitate yields 129 ATP
|
|
|
|
Fuel sparing during starvation
|
Glucose is needed for brain and RBC.
Protein is needed to sustain body function. Fat storage makes FAs available to sustain. |
|
|
|
Protein Sparing
|
During Prolonged Fast.
First, glycogen used. Then Protein metabolism reaches peak in 12 hours. However, after this point, less protein is metabolized and more FFA are metabolized. Tend to retain stored fat until needed in starvation |
|
|
|
Cell Membrane glucose transporters
|
Glycolysis requires glucose to enter cell membrane through transporters. Insulinm determines availability of transporters at cell plasma membrane.
|
|
|
|
FA and GLucose Metabolism
|
Share same endpoint (Acetyl CoA) so if excess FA there will be excess Acetyl CoA and glycolysis will shut down.
|
|
|
|
Metabolic Syndrome
|
One or more of:
Type II diabetes Impaired Glucose tolerance Insulin resistance + Two or more of: Hypertension Obesity Hypertricglyceremia Low HDL Microalbuminuria with often cooccuring: Hyperuricemia Hypercoagulability Hyperleptinemia yields Metabolic Syndrome leads to Diabetes, HIgh BP, Heart Disease, Stroke, Coagulopathies, Kidney Disease |
|
|
|
Quercetin
|
Flavonoid found in apple skins, berries, red wine, etc.
Helped mice run more by creating more mitochondria. FRS develops quercetin sports drink. Drink improves high intensity performance. Lance armstrong promotes No difference between placebo and Quercitin. Market Research is done to find "antidotes" to problems. |
|
|
|
Inborn errors of metabolism
|
Homocysteinuria
Phenylketonuria Tay-Sachs disease Maple Syrup Urine disease Tyrosinemia Gaucher's Disease Niemann-Pick disease Fabry's disease Glycogen storage diseases Galactosemia Hereditary Fructose Intolerance |
|
|
|
Metabolic Diseases of Muscle
|
Acid Maltase Deficiency(Pompe's disease)
Carnitine Deficiency Carnitine Palmityl Transferase Deficiency Debraucher Enzyme Deficiency Lactate Dehydrogenase Deficiency Myoadenylate Deaminase Deficiency Mitochondrial Myopathy Phosphofructokinase Deficiency Phosphoglycerate Kinase Deficiency Phosphoglycerate mutase deficiency phosphorylase deficiency (McArdle's Disease) |
|
|
|
Ratios of AMP/ADP/ATP
|
In liver vs muscle same ADP and AMP but ATP 3.0 in liver and 5.0 in muscle
|
|
|
|
anemia or ischemia
|
limits Oxygen delivery necessary for efficient fuel utilization
|
|
|
|
CHO processed into Glucose
|
all for circulation
|
|
|
|
FAs to Ketone Bodies
|
more easily transported around body (h20 soluble)
|
|
|
|
Glycerol, lactate, pyruvate
|
3 carbon intermediate molecule
glycerol is backbone to FA lactate and pyruvate come from FA metabolism |
|
|
|
Ethanol
|
2 carbon fuel
only used when built into FA |
|
|
|
Places TCA cycle occurs
|
Liver
Adipose Muscle Brain (not RBC) |
|
|
|
Places B-oxidation occurs
|
Liver
Muscle (not Adipose, Brain, RBC) |
|
|
|
Places Ketone Bodies Form
|
Liver
nowhere else |
|
|
|
Places Ketone Bodies Used
|
Some Adipose
Muscle Brain (when starved) (no rbc, no liver, no brain when free glucose) |
|
|
|
Place of Lipogenesis
|
Liver
Some adipose |
|
|
|
place of gluconeogenesis
|
Liver
nowhere else |
|
|
|
places of Glycogen metabolism
|
Liver
Muscle Some adipose Brain (sometimes) (no RBC) |
|
|
|
Lactate formed
|
Muscle during exercise
RBC (A little in brain, adipose, liver) |
|
|
|
B-Oxidation
|
break up long chain FAs into 2-carbon pieces
ketone bodies are end products of FA metabolism FAs not H20 soluble and dont transport across body well Ketone Bodies do |
|
|
|
Lipogenesis
|
make FAs out of excess fuel
|
|
|
|
Gluconeogenesis
|
processes glucose partway
produces glucose when a tissue needs it |
|
|
|
Glycogen
|
storage form of CHO
|
|
|
|
Lactate
|
last step in glycolysis
not usually used because products of glycolysis are taken into TCA if theres is low oxygen, lactate increases glycolysis muscle is primary lacatate formation site because energy demands are high and oxygen is not supplied fast enough |
|
|
|
Lipogenesis
|
make FAs out of excess fuel
|
|
|
|
Gluconeogenesis
|
processes glucose partway
produces glucose when a tissue needs it |
|
|
|
Glycogen
|
storage form of CHO
|
|
|
|
Lactate
|
last step in glycolysis
not usually used because products of glycolysis are taken into TCA if theres is low oxygen, lactate increases glycolysis muscle is primary lacatate formation site because energy demands are high and oxygen is not supplied fast enough |
|
|
|
Liver Energy
|
Uses everything but ketone bodies
|
|
|
|
Muscle energy
|
uses fat,ketone bodies, glycogen
|
|
|
|
RBC energy
|
uses glucose, processes it into lactate
|
|
|
|
Glycolysis decreases
|
as FFA become available
Glucose-6-Phosphate is first step in metabolizing glucose Amount of G-6-P indicates level of glycolysis FFA decrease, More G-6-P available FFA increase, less G-6-P |
|
|
|
CHO Pathway
|
CHO-->glucose--> either metabolism (glycolysis) or glycogen(storage)-->liver and muscles
|
|
|
|
Proteins Pathway
|
Proteins-->AAs--> MUscle(and other proteins) (no specific form of protein storage)
|
|
|
|
Fats Pathway
|
Fats-->FAs-->Triglycerides--> adipose
|
|
|
|
CHO Pathway Alternate
|
CHO-->glucose-->Acetyle CoA--> FAs-->triglycerides-->adipose
|
|
|
|
Stored fuels
|
adipose triglycerides>protein>glycogen
|
|
|
|
Circulating fuels
|
glucose>triglycerides>FFA
|
|
|
|
3 Effects FFA on Normal Cell
|
Decrease number of glucose transporters that come to surface and affect function of glucose transporters
decrease glucose transform to glycogen decrease glucose metabolism |
|
|
|
Kwashiorkor
|
Starvation
excceded capacity to using AAs via breaking down proteins\body makes use of fat (until no more fat, then make use of proteins) liver cant produce enough albumin which functions to maintain fluid inside bloodstream tissues are leaking and fluid collects in body cavities patient presents with swollen belly and stiff arms/legs |
|
|
|
Type II diabetes
|
Hyperglycemia: impaired glucose tolerance following a glucose load so glucose isnt going where it is supposed to
usually accompanied by insulin resistance associated with obesity possible mechanisms: fewer receptors, dysfunctional receptors, aberrant signaling, fewer or occluded glucose transporters |
|
|
|
ketogenic diet
|
helps epilepsy
|
|
|
|
Metabolic diseases
|
mostly autosomal recessive
|
|
|
|
Homocysteinuria
|
inability to process homocysteine
builds up in blood stream promotes clotting pectus excavatum is a marker children affected in utero are born with osteoporosis |
|
|
|
Phenylketonuria
|
phenylalanine hydroxylase that takes OH group that puts ring structure on tyrosine doesnt function properly
phenylalanine builds up, neurons function poorly |
|
|
|
Tay Sachs Disease
|
prevalent in jews of eastern european descent
defect in processing glycolipid that is prevalent in brain cells and it builds up children develop normally then regress and death is inevitable |
|
|
|
Mitochondria
|
outermost membrane is porous
inner membrane is thick and folded (cristae) holds proton gradient to use as energy cristae are studded with proteins for oxidative phosphorylation enzymes for TCA cycle are in matrix |
|
|
|
Mitochondrial Myopathies
|
Cause energy defects
|
|
|
|
Reproductive Hormones
|
testosterone
progesterone estradiol androgen prolactin |
|
|
|
Growth and Development Hormones
|
GH
TH Cortisol Growth Factors Prolactin |
|
|
|
Homeostatic Hormones
|
Aldosterone
VAsopressin Vitamin D Retinoic Acid (vitamin A) |
|
|
|
Energy Production, utilization, storage
|
insulin
glucagon epinephrine cortisol leptin |
|
|
|
RBC and Brain Metabolic Interactions
|
most fastidious and voracious organ needs constant glucose supply
|
|
|
|
Heart metabolic needs
|
FAs, lactate, ketone bodies
|
|
|
|
Muscle Metabolic Needs
|
FLEXIBLE
FAs major fuel source at rest Glucose from glycogen store is used @ earlier stage of exertion then FAs as dominant fuel |
|
|
|
Liver metabolic needs
|
Most flexible
Plays major role in keeping stable blood glucose level |
|
|
|
Intestine
|
digestion/absorption of nutrients
|
|
|
|
Blood
|
superhighway for balance/transportation of nutrients
|
|
|
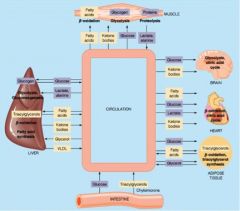
pathway of circulation
|
pathway
|
|
|
|
Exocrine Pancreas
|
Trypsinogen
Chymotrypsinogen Pancreatic Lipase Amylase |
|
|
|
Endocrine Pancreas
|
insulin (B cells)
glucagon (a cells) gastrin (Delta cells) pancreatic polypeptide (F cells) |
|
|
|
islet of langerhans
|
B cells in core
other cells mixed in mantle |
|
|
|
Insulin formation
|
preproinsulin-->proinsulin--> insulin
C-peptide is secreted with insulin (useful for diagnosis) interspecies insulin are similar |
|
|
|
Insulin formation
|
preproinsulin is secreted as random coil on membrane associated ribosomes with leader sequence--(assists with membrane transport)
leader sequence is cleaved after membrane transport resultant proinsulin folds into stable confirmation disulfide bonds form between alpha and beta chains connecting sequence connects alpha and beta chains and when cleaved alpha+beta chains+ disulfide bonds become a mature insulin molecule |
|
|
|
Insulin formation steps
|
1. nucleus (produce mRNA for preproinsulin production)
2. granular ER (synthesis of preproinsulin, which is cleaved by microsomal enzymes into proinsulin 3. Transfer vesicles to Golgi 4. Golgi (package, convert proinsulin to insulin) 5. Glucose into mitochondria, influx Ca++ 6. Secretory granules of insulin condense and are released |
|
|
|
Regulation of insulin step one
|
1. Glucose transporter 2 (GLUT2) is specific to liver and pancreas
responsible for glucose absorption senses blood glucose levels mediates transportation of glucose to B cells |
|
|
|
Regulation of insulin step two
|
increase glucose catabolism/atp synthesis
|
|
|
|
regulation of insulin step three
|
close K+ channels/open Ca++ channels
|
|
|
|
regulation of insulin step 4
|
Ca++ influx
|
|
|
|
regulation of insulin step 5
|
insulin synthesis/secretion
|
|
|
|
insulin signaling pathways for muscle and adipose tissue
|
Glucose transporter 4 (GLUT4) located in cytoplasm
translocates to plasma membrane with insulin stimulation |
|
|
|
insulin signaling pathways
|
1. insulin binds to insulin receptor
2. autophosphorylation of tyrosine on B subunits 3. phosphorylation of IRS (insulin receptor substrate) 4. GLUT4 translocates to Plasma membrane 5. Glucose influx 6. glycolysis 7. glycogen synthesis 8. FA synthesis |
|
|
|
Insulin effects on liver 1
|
increase glucose phosphorylation
|
affects glucokinase
|
|
|
insulin effects on liver 2
|
increase glycolysis
|
affects phosphofructokinase-1, pyruvate kinase
|
|
|
insulin effects on liver 3
|
increased glycogen synthesis
|
affects glycogen synthase
|
|
|
insulin effects on liver 4
|
increased FA synthesis
|
Affects AcetylCoA Carboxylase, ATP Citrate Lyase, Malic Enzymes
|
|
|
Insulin effects on liver 5
|
decreased gluconeogenesis
|
affects PEP carboxykinase, F-1,6-Bisphosphatase, Glucose-6-Phosphatase
|
|
|
insulin effects on liver 6
|
decreased glycogenolysis
|
affects glycogen phopshorylase
|
|
|
insulin effects on liver 7
|
increase pentose phosphate pathway
|
affects G-6-P dehydrogenase
|
|
|
insulin effects on adipose 1
|
increased glucose uptake
|
affects glucose carrier
|
|
|
insulin effects on adipose 2
|
increased glycolysis
|
affects phosphofructokinase-1
|
|
|
insulin effects on adipose 3
|
increased pentose phosphate pathway
|
affects Glucose-6-Phosphate Dehydrogenase
|
|
|
insulin effects on adipose 4
|
Increased Pyruvate Oxidation
|
affects pyruvate dehydrogenase
|
|
|
insulin effects on adipose 5
|
increased triglyceride utilization
|
affects lipoprotein lipase
|
|
|
insulin effects on adipose 6
|
increased triglyceride synthesis
|
affects glycero-3-phosphate acyl transferase
|
|
|
insulin effects on adipose 7
|
decreased lipolysis
|
affects hormone sensitive lipase
|
|
|
insulin effects on skeletal muscle
|
increased glucose uptake
|
affects glucose carrier
|
|
|
insulin effects on skeletal muscle
|
increased glycolysis
|
affects phosphofructokinase-1
|
|
|
insulin effects on skeletal muscle
|
increased glycogen synthesis
|
affects glycogen synthase
|
|
|
insulin effects on skeletal muscle
|
decreased glycogenolysis
|
affects glycogen phosphorylase
|
|
|
insulin effects on skeletal muscle
|
increased protein synthesis
|
affects translation initiation complex
|
|
|
Biochemical affects of insulin
|
increased cell permeability to glucose (muscle and adipose)
increased glycolysis increased glycogen synthesis increased triacylglycerol synthesis decreased gluconeogenesis decreased lipolysis decreased protein degradation increased protein, DNA, RNA synthesis |
|
|
|
Physiological affects of insulin
|
signals fed state
decreased blood glucose level increased fuel storage increased cell growth and differentiation |
|
|
|
Glucagon
|
synthesized in alpha cells in pancreas
preproglucagon-->proglucagon--> glucagon single chain 29 polypeptides secretion is inhibited by glucose and insulin increased secretion stimulated by: AAs, catecholamines, glucocorticoids, nervous system |
|
|
|
receptor mediated activation of PKA
|
1 molecule glucagon-->
~20 G-Proteins--> ~100 cAMP--> ~100-1000 phosphorylations--> net effect~10^5, 10^6 |
|
|
|
Glucagon effects on blood glucose levels
|
glucose decreased, then glucagon increases, cAMP increases, PKA increases
increased PKA leads to increased phosphorylase--> increased glycogenolysis--> increased glucose increased PKA leads to decreased glycogen synthase--> decreased glycogen synthesis --> increased glucose increased PKA leads to decreased pyruvate kinase--> increased gluconeogensis--> increased glucose increased PKA leads to decreased PFK2, increased F2,6BPase--> decreased F2,6BP--> decreased glycolysis--> increased glucose |
|
|
|
glucagon increases
|
increases glucagon/glucagon receptor--> increased cAMP--> increased PKA--> increased phosphorylation-->decreased glycogen synthase--> decreased glycogen synthesis -->increased glycogen phosphorylase--> increase glycogen degradation--> increased glucose
|
|
|
|
decreased glycolysis
|
affects
glucokinase (induction/repression) PFK1 (other) Pyruvate Kinase (induction/repression) |
|
|
|
Increased gluconeogenesis
|
affects
PEP carboxylase (induction/repression) Fructose 1,6 bisphosphatase (induction/repression, other) glucose-6-phosphatase (induction/repression) |
|
|
|
decreased glycogen synthesis
|
affects glycogen synthase (phosphorylation)
|
|
|
|
increased glycogenolysis
|
affects glycogen phosphorylase (phosphorylation)
|
|
|
|
decreased FA synthesis
|
affects AcetylCoA carboxylase (induction/repression) (phosphorylation)
|
|
|
|
increased FA oxidation
|
affects carnitine palmityl transferase-1 (induction/repression)
|
|
|
|
High CHO meal
|
increased insulin, decreased glucagon
Fructose-6-P --> Fructose 2,6-P phosphorylated by PFK2 |
|
|
|
Fasting
|
decreased insulin, increased glucagon
fructose 2,6-p-->Fructose-6-P dephosphorylated byF-2,6BPase |
|
|
|
Glucose metabolism
|
Glucose--> Fructose-6-Phosphate
fructose-6-phosphate + PFK1(from F2,6BP)--> Fructose 1,6 BP--> 2 pyruvates Fructose 1,6 Bisphosphate +F-1,6-BPase(from F 2,6BP)--> Fructose-6-Phosphate |
|
|
|
type I diabetes
|
childhood, thin, decreased insulin, increased glucagon, increased gluconeogenesis, increased blood glucose
insulin is remedy 10% cases |
|
|
|
Type II diabetes
|
adolescence, obese, 90% cases
insulin receptor downregulation glucagon is similar nutrient usage is down increase in blood glucose/cholesterol/FA treat with exercise and dietary modifications |
|
|
|
catecholamines neurotransmitter
|
DOPA
dopamine |
|
|
|
catecholamines hormone
|
Norepinephrine (transmitter)
Epinephrine |
|
|
|
tyrosine anabolism
|
tyrosine + tyrosine hydroxylase (rate lmiting) --> DOPA + DOPA decarboxylase--> Dopamine + Dopamine hydroxylase--> Norepinephrine + Norepinephrine n-methyltransferase--> Epinephrine
|
|
|
|
synthesis in adrenal medulla
|
physical exertion/psychological stress/cold---> anterior pituitary
hypothalamus--> anterior pituitary (catecholamine releasing)--> adrenal medulla |
|
|
|
signal transduction of B2 receptors
|
identical to glucagon receptor
Activate adenylate cyclase--> increas in cAMP--> activate PKA |
|
|
|
catecholamines
|
slows down gut
speeds up heart increase sweating mobilize stores of energy |
|
|
|
epinephrine effect on liver
|
Increase EPI--> adrenergic receptor--> increase cAMP--> increase in PKA-->
increase gluconeogenesis decrease glycolysis increased glycogenolysis decrease glycogenesis --> increase blood glucose opposite effect of epi on heart muscle vs liver |
|
|
heart
|
heart
|
|
|
|
effect of epi on adipose
|
increase lipolysis
hormone sensitive lipase |
|
|
|
effect of epi on adipose
|
decreased triglyceride utilization
lipoprotein lipase |
|
|
|
effect of epi on liver
|
decreased glycolysis
phosphofructokinase-1 |
|
|
|
effect of epi on liver
|
increased gluconeogenesis
fructose-1,6-bisphosphatase |
|
|
|
effect of epi on liver
|
decreased glycogen synthesis
glycogen synthase |
|
|
|
effect of epi on liver
|
increased glycogenolysis
glycogen phosphorylase |
|
|
|
effect of epi on liver
|
decreased FA synthesis
lipoprotein lipase |
|
|
|
effect of epi on heart
|
increased glycolysis
phosphofructokinase-1 |
|
|
|
effect of epi on heart
|
decreased glycogen synthesis
glycogen synthase |
|
|
|
effect of epi on heart
|
increased glycogenolysis
glycogen phosphorylase |
|
|
|
effect of epi on heart
|
increased triglyceride utilization
lipoprotein lipase |
|
|
|
cortisol/glucocorticoids
|
chronic stress--> corticotropin releasing factor (CTRF) from hypothalamus--> ACTH (adrenocorticotropic hormone) from anterior pituitary gland--> cortisol and glucocorticoids (from adrenal cortex)--> glucocorticoid receptor--> gene expression -->long term--> increased lipolysis, increased protein degradation, increased gluconeogenesis
|
|
|
|
Cortisol/Glucocorticoids Effects Adipose
|
Increase Lipolysis
affects hormone sensitive lipase |
|
|
|
Cortisol/Glucocorticoids Effects muscle
|
Increase protein degradation
|
|
|
|
Cortisol/Glucocorticoids Effects liver
|
increase glycogen synthesis, increased gluconeogenesis
affects glycogen synthase enzymes in amino acid metabolism PEP carboxykinase |
|
|
|
Low Blood Glucose Cascade
|
LBG-->Pancreas (A cells)--> glucagon
LBG-->Hypothalmic regulatory center-->ANS-->norepi ANS--> adrenal medulla--> EPI LBG-->hypothalmic regulatory center-->pituitary-->ACTH--> Adrenal cortex--> cortisol |
|
|
|
Leptin
|
16kDa peptide hormone from adipocyte. amount proportional to bodyfat
+ leptin=when need tolose weight - leptin=when need to gain weight function to decrease food intake (satiety) and increase energy expenditure (metabolism) |
|
|
|
Db (leptin receptor)
|
membrane protein enriched in hypothalamus. Coded for by Db gene (diabetes gene). Without this gene, no receptors, and become fat
|
|
|
|
parabiosis
|
circulatory systems of two mice joined together
Ob with normal-->lose weight Db with normal--> normal lose wt Ob with Db--> Ob lose wt |
|
|
|
leptin therapy
|
used with people with leptin deficiency. no effect in those without deficiency
|
|
|
|
orexigenic
|
appetite stimulating
NPY producing AgRP producing GHRELIN (stomach) |
|
|
|
Anorexigenic
|
Appetite suppressing
POMC producing (a-MSH) PYY intestine CCK intestine Leptin adipose Insulin pancreas |
|
|
|
NPY
|
neuropeptide Y
|
|
|
|
AgRP
|
Agouti-related peptide
|
|
|
|
POMC
|
propriomelanocortin
|
|
|
|
a-MSH
|
alpha melanocyte stimulating hormone
|
|
|
|
Vitamin D and Vitamin A
|
can penetrate cell membrane and act on receptor in nucleus
|
|
|
|
Cortisol
|
can penetrate cell membrane
|
|
|
|
B-oxidation
|
FAs precursors to acetyl coA and used to produce ketone bodies
|
|
|
|
zymogens
|
prevent digestion of pancreas itself
|
|
|
|
pancreatic enzymes
|
trypsinogen, chymotrypsinogen, pancreatic lipase, amylase
|
|
|
|
Dela cells
|
create gastrin and somatostatin
|
|
|
|
F cells
|
pancreatic polypeptides and inhibits somatostatin
|
|
|
|
insulin
|
signal peptide (lead strand) removed by signal peptidase
|
|
|
|
procine or bovine insulin
|
used in humans
|
|
|
|
Glucose
|
directly signals beta cells to release insulin
|
|
|
|
GLUT2
|
found in B cells, liver
senses blood glucose levels enhances glucose influx causes increased glycolysis and ATP synthesis increased ATP closes K+ channels, K+ no longer flows into cell indirectly opens CA++ channel causes direct release of insulin (microfilaments contract in response to CA++ and will release vesicles of insulin into blood stream CA++ binds to calmodulin--> transcription regulation of insulin through calcium response element binding protein CREB + Ca++ has bound insulin gene promoter enabling transcription of insulin gene for next round of secretion |
|
|
|
Tetramer GLUT2
|
four subunits (2 alpha, 2 beta
transciption of these subunits controlled by one promoter (so 1:1 ratio) a-subunits extracellular and bind insulin B subunits cross membrane -c-terminus is in cytosol and equipped with tyrosine kinase activty insulin binds receptor--> autophosphorylation of c-terminal B subunits--> phosphorylation of IRS--> glucose transporter recruited to plasma membrane |
|
|
|
GLUT4
|
found in muscle and adipose tissue
normally located in cytosol translocates to cell membrane with insulin stimulation causes-glucose influx catabolism muscle glycogen made in muscle, FAs in adipocytes, stored as triacylglycerol |
|
|
|
glucagon
|
inhibited by glucose/insulin
stimulated by AAs and their derivatives (arginine, alanine, GABA, catecholamines, glucocorticoids, GI hormones, increased FFA, nervous system control carried to target tissue through bloodstream G-protein coupled receptor stimulation--> G-protein activation-->activate adenylate cyclase-->cAMP + PKA--> protein phosphorylation PKA has catalytic and regulatory domains cAMP binds to and removes regulatory domains |
|
|
|
GPCR
|
G-protein coupled receptor uses GTP to make G-protein release GDP and bind a GTP
adenylate cyclase uses ATP to generate cAMP requires 4 cAMP to fill regulatory domains of PKA because there are 2 domains with 2 binding sites each phosphodiesterase turns cAMP into AMP to reduce cAMP concentration -without cAMP, the regulatory domain of PKA will bind the catalytic domain to stop the reaction strength of the signal depends on how many glucagon bind receptors and how high cAMP concentration gets |
|
|
|
glycolysis
|
uses glucose to generate pyruvate and ATP (regulated by Fructose-2,6-Bisphosphate (f-2,6-BP) which activates PFK-1 and inhibits Fructose 1,6-Bisphosphatase (F-1,6-BPase) thereby inhibiting the reverse process
|
|
|
|
PFK-2
|
increases F-2,6-BP
|
|
|
|
F-2,6-BPase
|
decrease F-2,6-BP
|
|
|
|
glucagon
|
leads to phosphorylation, activates F-2,6-BPase
|
|
|
|
insulin
|
leads to dephosphorylation, activates PFK-2, PFK-1
|
|
|
|
GLUT1 and GLUT 3
|
RBC and Brain, high affinity, low capacity
|
|
|
|
GLUT 2
|
liver, pancreas, low affinity, high capacity
|
|
|
|
GLUT4
|
adipose and muscle (only if insulin is present)
increase glucose levels leads to glycosylation -DNA, RNA, etc. glycosylation can modify transcription and translation of proteins increased glucose all the time can be toxic by having ttransporters, glucose is taken into cell in a way that is not toxic |
|
|
|
Catecholamine synthesis
|
Tyrosine +tyrosine hydroxylase--> DOPA+dopamine decarboxylase (removes carboxyl group in form of CO2)--> Dopamine + domanine hydroxylase (with Vit C as a cofactor adds OH group)--> Norepinephrine +n-methyltransferase (adds methyl to NH2)--> epinephrine
|
|
|
|
catecholamines
|
work like glucagon but faster
in heart increases F-2,6-BP in liver Decreases F-2,6-BP (want gluconeogenesis in liver) |
|
|
|
stress pathway
|
stress-->hypothalamus--> CRF--> anterior pituitary--> ACTH--> adrenal cortex--> cortisol/glucocorticoids-->
lipolysis, gluconeogenesis, protein degradation |
|
|
|
leptin
|
deletion of OB gene leads to obesity
Db knockout grows bigger than Ob knockout |
|
|
|
choleocystikinin
|
released from small intestine during eating to promote sense of fullness and releasing of bile and digestive enzymes
|
|
|
|
ATP Phosphate bonds
|
gamma = high energy
B=less high a=never used |
|
|
|
number of mitochondria
|
depends on energy needs
~50% of cytoplasm of cardiac cells occupied by mitochondria mature RBC has no mitochondria |
|
|
|
mitochondria
|
outer permeable membrane
intermembrane space contains cytoplasm inner membrane space is not permeable (including protons) inner membrane folded into cristae (surface area) inner membrane compartment called matrix |
|
|
|
mitochondrial matrix
|
contains
TCA cycle enzymes B oxidation ATP synthase/ATP ETC Mitochondrial DNA |
|
|
|
B oxidation
|
makes 2-carbon Acetyl CoA
|
|
|
|
Complex
|
group of functional units (protein)
|
|
|
|
electron transport systems
|
complexI-->coQ-->complexIII-->complexIV
complexII-->coQ-->complexIII-->complexIV I and II merge through coQ |
|
|
|
proton pump efficiency
|
10 protons pumped to intermembrane space when begin at complex I
6 when begin at complex II |
|
|
|
COmplex I
|
NADH + H+ --> NAD+ (nadh is reducing equivalent)
|
|
|
|
Complex II
|
succinate-->fumarate
(TCA cycle has 8 steps/enzymes, but the only substrate on membrane of TCA cycle is succinate dehydrogenase) Complex II is not transmembrane |
|
|
|
protons pumped
|
complex I-->4
complex II-->0 complex III-->2 Complex IV--> 4 |
|
|
|
complex I
|
>40 subunits
MW=1MDa 1 FMN (similar to FAD, extracts 2H from NADH to give to CoQ) 8 Fe-S clusters (accept e- from NADH), donate e- to Fe-S and then CoQ Fe serves as a transport of electrons Gaps b/t subunits form channel for proton pumping alternate name (NADH dehydrogenase) inactivated by rotenone, riboflavin deficiency |
|
|
|
complex II
|
alt name: succinate dehydrogenase
has FAD and Fe-S succinate donates e- to CoQ inactivated by malonate |
|
|
|
complex III
|
CoQ/CoEnzymeQ/Ubiquinone
-has CoQ -NADH dehydrogenase donates electrons to bc1 complex inactivated by generation of free radicals, doxorubicin cytochrome bc1 complex -ubiquinone-cytochrome c oxidoreductase has Fe-S, Hemes (b562, c-1) inactivated by antimysin, demerol, Fe deficiency cytochrom c has Heme C bc1complex donates e- to cytochrome oxidase inactivated by Fe deficiency |
|
|
|
Complex IV
|
cytochrome oxidase
complex IV cytochrome AA3 has Heme-a cytochrome c-->O2 inactivated by carbon monoxide, cyanide, ischemia, Fe and Cu deficiency (leads to Fe deficiency) Carbon monoxide (binds to heme, blocks O2) |
|
|
|
Heme
|
Fe molecule in center
|
|
|
|
cyanide
|
binds to heme Fe in complex IV prevents O2 from being used as a transporter and binding to heme
|
|
|
|
general cycle
|
AH2 (substrate) oxidized-->A + NAD+--> NADH
Fp(flavoprotein) +NADH--> FpH2 and NAD+ FpH2 + 2 Fe3+--> Fp + 2 Fe2+ 2Fe2+ + 1/2 O2--> H20 1. reduced fuel passes electron to coenzyme 2. the electrons are passed from the coenzyme to a flavoprotein in a complex 3. the flavoprotein passes them to an iron containing cytochrome group 4. the last cytochrome passes them to oxygen, reducing it to water |
|
|
|
electrochemical potential
|
cytosolic side has + charge (acidic), matrix side is - charge (basic) because of proton pump
driving force of ATP synthesis a. electro-membrane potential b. chemical-involves proton particles c. electrochemical potential |
|
|
|
ATP synthase
|
a protein complex
H+ channel -Fo (C-subunits) Catalytic Domain -F1(a,b,a,b,y,d) use the electrochemical potential to synthesize ATP C-complex embeds in membrane -flow of protons turns complex -each c-subunit moves 30 degrees/proton. need 12H+ to turn 360 degrees Y turns 360, 3 ATP synthesized pool of protons inside the matrix are used for pumping |
|
|
|
oxidative phosphorylation
|
complex I- FMN is main acceptor
complex II- FAD is main acceptor |
|
|
|
mitochondrial inner membrane transporters
|
Adenosine Nucleotide Translocase (antiporter)
-ADP--> matrix -ATP--> intermembrane space charge difference gives process movement (electrochemical potential) ATP synthase (uniport) -H+--> matrix Phosphate translocase (symporter) H2PO4--> matrix H+--> matrix concentration gives movement (chemical potential) |
|
|
|
mitochondria ATP synthesis
|
oxidation of NADH or succinate
-electron passed to O2 through ETC to form H20 -formation of electrochemical potential -potential used by ATP synthase to synthesize ATP -oxidative phosphorylation -oxidation and ATP synthesis |
|
|
|
different efficiencies of electron transfer chains
|
complex I extracts elctrons from NADH and transfers them to complex III (and pumps 4H+)
complex II extracts electrons from succinate and passes them to complex III (no pump H+) Electron transfer from complex III to complex IV pumps 2H+ Electron transfer from complex IV to O2 (pumps 4H+) |
|
|
|
ATP synthesized
|
NADH oxidation pumps 10H+, synthesizes 2.5 ATP
FADH2 oxidation pumps 6H+ and synthesizes 1.5 ATP |
|
|
|
Vitamin B3 derivatives
|
NAD+ (nicotinamide adenine dinucleotide)
NADP+ (nicotinamide adenine dinucleotide phosphate) |
|
|
|
Vitamin B3 deficiency
|
glossitis (swollen tongue, inflamed)
pellagra (diarrhea, dermatitis, dementia) B3 overdose is flushing |
|
|
|
Vitamin B2 derivatives
|
FMN (flavin mononucleotide)
FAD (flavin adenine dinucleotide) |
|
|
|
Vitamin B2 deficiency
|
cheilosis (inflamed lips, scaling/fissures at corner of mouth, glossitis)
corneal vascularization |
|
|
|
ATP synthesis and utilization
|
Glucose, FAs, AAs--> acetyl CoA--> TCA cycle
FA is more efficient than glucose and proteins (more easily removed 2-carbon groups Ketone bodies and Ethanol are energy enriched (easy to produce Acetyl CoA) glucose-->pyruvate-->TCA-->FADH2 NADH-->ATP |
|
|
|
creatine phosphate
|
energy can be stored temporarily as this
|
|
|
|
Malate-Aspartate shuttle
|
(antiporter)
two enzymes -malate dehydrogenase -aspartate aminotransferase two transporters -malate a-ketoglutarate -glutamate aspartate transporter H is transferred to oxaloacetate-->Malate which helps move NADH from cytosol into matrix by using a reducing equivalent) (enzymes catalyze forward and backward reaction) regenerates NADH |
|
|
|
glycerol-3-phosphate shuttle
|
DHAP (dihydroxyacetone phosphate) + Glycerol-3-Phopshate dehydrogenase--> Glycerol-3-Phosphate
does not regenerate NADH--start at CoQ |
|
|
|
ATP in respiratory control
|
"pulls" demand of O2 forward
|
|
|
|
oligomycin inhibits ATP synthase
|
binds to Fo of ATP synthase
blocks proton channel inhibits atp synthesis -lower atp, higher adp etc is enhanced increased electrochemical potential ultimate ETS stops keeps proton gradient intact slows 02 consumption |
|
|
|
uncoupling phosphorylation from electron transport
DNP |
DNP (2,4 dinitrophenol) destroys H+ gradient allows continued ADP stimulation of 02 consumption without ATP synthesis
it is a weak acid/hydrophobic DNP is protonated in the intermembrane space protonated DNP diffuses into matrix DNP is deprotonated in matrix H+ gradient is disrupted no ATP synthesized |
|
|
|
Thermogenin
|
protein complex like DNP
oxidation of NADH/FADH formation of H+ gradient H+ gradient uncoupling by thermogenin generation of heat reducing ATP synthesis |
|
|
|
ADP
|
required for ATP synthesis
atp hydrolysis controls ATP synthesis ADP effects -enhances oxidative phosphroylation -increases rate of TCA cycle allosterically -NADH is required for ATP synthesis -NAD+ is required for TCA cycle energy needs which consume NADH allow NAD+ to increase rate of TCA cycle As energy needs decline, excess NADH slows down TCA cycle rations of ATP/ADP and NADH/NAD control rate of activity |
|
|
|
interlocking regulation
|
glycolysis
pyruvate oxidation citric acid cycle oxidative phosphorylation all regulated by relative concentrations of AMP ADP ATP NAD NADH |
|
|
|
calorie restriction/NADH/longevity
|
CR seems to increase lifespan
during CR, Sirt1 (deacetylase) activity increases NAD+ serves as Sirt1 activator increased ratio NAD/NADH lowers cholesterol, fasting glucose, blood pressure slows aging, increases longevity resveratrol similar effects as CR without CR |
|
|
|
muscle work
|
myosin heads engage with actin at multiple sites
myosin head has an ATP binding domain and is an ATPase changes in myosin head conformation upon binding ATP, hydrolyzing ATP, or realeasing ADP allows contact with Actin and slide actin filament forward ATP-releasing Y, B phosphate bonds provide energy |
|
|
|
Increase ATP
Increase ADP |
body has energy/body needs anergy
|
|
|
|
1,3 Bisphosphoglycerate
Phosphoenol Pyruvate |
have PO4 bonds, high energy, that can be made into ATP
importnat in glycolysis, substrate level phosphorylation in RBC (no mitochondria) |
|
|
|
acetyl CoA
|
all fuel ends up as acetyl CoA
has high energy ester bond |
|
|
|
creatine phosphate
|
phosphate becomes sequestered
after big meal, some ATP is stored as creatine phosphate in muscles releases more energy than just ATP ATP lasts for 2 seconds, Creatine PO4 kicks in for 8 seconds, then muscle dips into glycogen all high energy molecules can interconvert into ATP also participate in energy requiring reactions |
|
|
|
energy carrier molecules
|
NAD/NADH
NADP/NADPH FAD/FADH2 |
|
|
|
oxidative phosphorylation
|
rxn by which ATP synthesis occurs
most efficient ATP reduction (O2 reduced) |
|
|
|
electrochemical gradient
|
protonmotive force
|
|
|
|
electron transport chain
|
atp synthase (Fo/Fi ATPase)
|
|
|
|
fuels used to produce reducing equivalent and ATp
|
at substrate level phosphorylation
fuels being oxidized, O2 reduced to H20 |
|
|
|
BMR
|
weight in kg x 24
|
|
|
|
brain's need for glucose is driving force in energy metabolism
|
liver regulates the concentration of fuels in blood
-disposes of excess fuel from diet -provides fuels in between meals -interconverts fuels to glucose and keto acids so brain has enough fuel |
|
|
|
brain
|
requires supply of fuel and O2
cannot burn fatty acids uses 20-25% REE |
|
|
|
muscle
|
significant CHO reserves as glycogen
utilizes glucose or fat fuels major reserve of body protein spares brain fuels during exercuse |
|
|
|
RBC
|
only glucose, lactate
|
|
|
|
kidney
|
uses ATP to excrete wastes excretes nonvolatile wastes
excretes excess acid |
|
|
|
lungs
|
excretes volatile wastes (CO2)
|
|
|
|
coupling energy requiring reactions with energy releasing reactions
|
endergonic with exergonic
balance required globally in cell or organism |
|
|
|
synthetic operations coupled to energy production
|
reflects sum of individual coupled reactions
|
|
|
|
compare metabolic situations
|
fed vs fasted vs starved
|
|
|
|
type I diabetes
|
no insulin
|
|
|
|
type II diabetes
|
insulin has little effect
|
|
|
|
Alcoholism
|
increases NADH
|
|
|
|
insulin
|
stimulates glucose to be transported into cell
activates phosphatase which activates pyruvate kinase activates glycogen synthases inactivates glycogen phosphorylase inactivates glycogen phosphorylase kinase |
|
|
|
GLUT 4
|
muscle and adipose
|
|
|
|
GLUT 2
|
High Km, low affinity
liver induces glucokinase through insulin have to have insulin around before liver will take it in |
|
|
|
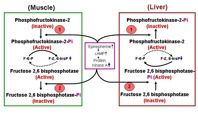
Fructose 2,6 Bisphosphate
|
made in response to insulin
activated by PFK1 activates glycolysis (not in any pathways) |
|
|
|
glycogen
|
fasted state
break down liver glycogen send glucose into blood stream activates protein kinase A induces gluconeogenic enzymes F2,6BP synthesis is inhibited (PFK2 is phosphorylated) cAMP activated protein kinase is stimulated to inhibit PK activates gluconeogenic pathway and shuts off glycolysis PKA phosphorylates GLYCOGEN SYNTHASE and GLYCOGEN PHOSPHORYLASE KINASE but only activates glycogen phosphorylase kinase to avoid a futile cycle |
|
|
|
TCA cycle
|
regulated by high NADH and ATP levels (both turn off this cycle)
Insulin activates pyruvate dehydrogenase (PDH) phosphatase and thereby activates the PDH complex PDH turns pyruvate into Acetyl CoA turning on the TCA cycle |
|
|
|
proteins
|
insulin makes some AAs go into tissues easier
|
|
|
|
FA synthesis
|
glycolysis is going (with lots of insulin)
phosphatase activates Acetyl CoA Carboxylase Insulin stimulates synthesis and secretion of lipoprotein lipase (LPL) this breaks down triglycerides into FAs for storage (LPL activated by apoCII) insulin induces FA synthesis enzymes -citrate lyase -AcCoA carboxylase -FA synthase insulin also induces NADPH synthesis, malic enzyme and G6PDH |
|
|
|
FA breakdown
|
bring FAs into liver so they can be broken down to make KBs
PKA is important stimulating hormone sensitive lipase (HSL) PKA inactivates Acetyl CoA carboxylase |
|
|
|
Liver
|
donates NADH to ETC to make energy
HOMEOSTASIS -carbohydrates glycolysis/TCA gluconeogenesis storage of glycogen for glucose production -AAs liver uses all AAs but BCAA urea cycle to remove extra ammonia -Lipids synthesis and secretion of bile acids and salts beta oxidation KB production FA synthesis VLDL packaging and secretion HDL packaging and secretion Uptake of chylo remnants Uptake of HDL SYNTHESIS/PROCESSING PRoteins -Heme synthesis in liver (and other tissues) bilirubin concentration -circulating proteins synthesis albumin haptoglobin, ceruloplasm blood clotting factors clotting inhibitors acute phase proteins -CYP450 system and oxidation reactions ETOH oxidation drug oxidation -storage vitamins cholesterol iron and other metals -EXCRETION/FILTRATION drugs after CYP450 bilirubin after conjugation poisons from gut DEFENSE excretion of IgA (against bacteria in gut) Macrophages (VonKupffer cells) and phagocytose bacteria |
|
|
|
Alcohol
|
increases NADH concentration
-alcohol dehydrogenase and aldehyde dehydrogenase both generate NADH ETOH+alcoholDH--> acetaldehyde + NADH --> acetate goes into muscle to be used to form Acetyl CoA for the TCA cycle ACS(Acetyle CoA synthetase) to use acetate for energy membrane fluidity is altered and can lead to toxic effects in brain acetaldehyde forms products with other proteins acetate gives high amounts of acetyl CoA which leads to increased FA synthesis (steatosis in alcoholics) increases NADH/NAD ration will increase the amount of lactate going to pyruvate (inhibits gluconeogenesis and TCA cycle) high NAD also inhibts FA B-oxidation increases glycerophosphate (precursor to TG formation) leads to increase TG in liver metabolism ETOH generates high NADH/NAD ratio high NADH/NAD ratio inhibits FA B-oxidation and TCA cycle (FA accumulate) FA reesterified to TG because high NADH/NAD ratio generates Glycerol-3-P from DHAP High NADH/NAD ratio inhibits TCA cycle (oxaloacetate goes to malate) High NADH/NAD ratio shifts OAA toward malate, acetyl CoA goes toward KB formation cannot do gluconeogensis because pyruvate goes toward lactate because of high NADH/NAD ration (lactic acidemia) Uric acid competes with other acids (lactic, salicylic, etc) for transport into kidney (break down DNA by acetaldehyde) gluconeogenic precursor AAs (via pyruvate) not used for GNG because push towards lactate alanine will go to pyruvate, pyruvate will go to lactate (causes hypoglycemia) NADH are blocking PDH. so OAA is going toward malate which goes into cytosol and stops gluconeogenesis |
|
|
|
Type I db
|
hormonal (no insulin, lots of glucagon)
|
|
|
|
type II db
|
hormonal, lots of insulin, does not work correctly, (no glucagon)
|
|
|
|
glycolysis
|
not much glucagon, lots of insulin
glucokinase is inducible (high Km) PFK1 is active when low ATP, high AMP and high F2,6BP PFK1 is inactive when high ATP, low AMP, high F 2,6BP pyruvate kinase is active with insulin and inactive with glucagon |
|
|
|
gluconeogenesis
|
low insulin, high glucagon levels
G-6-Phosphate is inducible with cortisol and cAMP via glucagon Pyruvate carboxylase is activated by Acetyl CoA PEP carboxykinase is inducible and activated by cAMP (via glucagon) |
|
|
|
FA oxidation
|
fasted state, increase FA/TG from adipose (via HSL)
malonyl CoA inhibits newly synthesized FA transport into mitochondria via CPTI mitochondria keeps compartments separated (all synthesis in cytosol) |
|
|
|
FA synthesis
|
fed state, increase FA/TG from digestion, increase ATP, NADPH, Acetyl CoA, citrate (tca)
citrate activates Acetyl CoA carboxylase and is used to make Acetyl CoA Cytosol - Malonyl CoA inhibits movement of acyl group into mitochondria |
|
|
|
TG breakdown
|
no futile cycle because must make glycerol-3-phosphate from glucose and if there is no glucose there will be no TG synthesis
HSL breaksdown TG into FA to be used for energy. HSL controlled by glucagon (activate) insulin (inhibit) TG breakdown by LPL to release FA from VLDL and chylomicrons for storage in adipose or energy in muscle |
|
|
|
TG synthesis
|
TG not synthesized from glycerol + FFA because no glycerol kinase in adipose tissue (most get from glucose)
TG only synthesized from glycerol derived from glucose TG synthesis happens in liver |
|
|
|
glycogen breakdown
|
glycogen phosphorylase is going to be active when you are breaking glycogen down because of the phosphate group that is on it
it will be inactive when dephosphorylated by a phosphatase gylcogen phosphorylase will be active when there is low insulin and high glucagon because the body needs energy and breaks down glycogen |
|
|
|
glycogen synthesis
|
glycogen synthase is active during synthesis
-dephosphorylated by phosphatase (to activate) inactivated by PKA glycogen synthase will be active when insulin is high and glucagon is low because the body has excess energy sources and can store it |
|
|
|
urea cycle on or off
|
protein synthesis or proteolysis
feeds forward because there are a lot of AAs so the body wants to get rid of some of those nitrogens by putting them on Urea Carbamoyl P synthetase -CPSI allosterically inactivated by N-Acetyl glutatmate (NAG) NAG not found in cycles only used to activate first step in urea cycle (NAG synthesized from glutamate and activated by arginine) urea cycle enzymes are under induction or repression of synthesis (high protein diet vs. prolonged fasting) |
|
|
|
biotin
|
carboxylation reactions
pyruvate carboxylase (pyruvate-->OAA) acetyl CoA carboxylase(acetyl coA-->malonyl) propionylCoA carboxylase post-translational carboxylases |
|
|
|
B1 Thiamine
|
TPP
cofactor in oxidative decarboxylation of alpha-keto acids -pyruvate dehydrogenase -alpha-ketoglutarate dehydrogenase -BCAA dehydrogenase formation or degradation of alpha-ketols by transketolase |
|
|
|
B2 Riboflavin
|
redox/oxidation reaction
used in TCA cycle and Beta-oxidation biologically active as FMN and FAD |
|
|
|
B3 Niacin
|
NAD/NADP are co-enzymes in redox reactions. accepts a hydride ion
|
|
|
|
B5 pantothenic acid
|
CoA
transfers acyl groups via a thiol group as activated thiol esters (CoA) |
|
|
|
B6 pyroxidine
|
PLP
decarboxylation transamination synthesis (heme): glycine (AA) is decarboxylated AA decarboxylation to form neurotransmitters (GABA, 5HT, histamine) transamination reactions a step in tryptophan degradation |
|
|
|
B12 cobalamin
|
nucleotide synthesis
transfers methyl group to form methionine involved in pyrimidine nucleotide synthesis methylmalonyl coA-->succinyl CoA (methylmalonyl coA mutase) carbons from val, ile, thr, thymine, last 3C of odd chain FA-->succinyl CoA odd chain FA metabolism and BCAA metabolism |
|
|
|
folic acid
|
receives 1C fragments from donors(ser, gly, his, etc( and transfers to intermediates in synthesis of AA, purines, thymidine
one carbon metabolism |
|
|
|
C(ascorbic acid)
|
antioxidant, free radical scavenger in aqueous environment
hydroxylation of prolyl residues on collagen by prolyl hydroxylase in preparation for cross-linking |
|
|
|
d (sterols)
|
hormone like functions to maintain CA2+ levels
increases uptake Ca++ by intestine minimizes loss Ca++ by kidney stimulates bone resorption if necessary |
|
|
|
E (alpha-tocopherol)
|
antioxidant
lipophilic free radical scavenger protects against lipid peroxidation |
|
|
|
K
|
modify clotting factors II, VII, IX, X
carboxylation of glutamate to gamma-carboxy-glutamate |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
inborn errors of metabolism
|
rare genetic diseases
blockage in metabolic pathway lower activity/complete deficiency of specific enzyme substrate will accumulate, product will be low substrate will then take other pathways accumulate in tissues and organs and become toxic and harmful pattern of inheritance -can be young/infant onset or adult higher severity with younger onset (dtect disease with pheotypic expression) milder with adult onset |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
AA diseases (PKU) organic acids, galactosemia, urea cycle disease
|
most common IBM
1/4200 PKU is most common |
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Lysosomal storage disease
|
1/12000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
Peroxisomal disorder
|
1/30000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
respiratory chain-based mitochondrial disease
|
1/33000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
glycogen storage disease
|
1/43000
|
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
lysosomes
|
cellular organelles that recycle macromolecules (proteins, glycoproteins, lipids, phospholipids, sphingolipids)
break down for reuse ~70 enzymes in lysosomes optimal activity at pH=5 |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
lysosomal storage disorders
characteristics |
defect can be at particular reaction site of lysosomal enzyme or coenzyme of reaction or posttranslational modification protein or lysosomal transport protein
results in accumulation of substrate in lysosome more than 5 different LSDs most autosomal recessive (Hunter and Fabry's are X linked) onset typically in infancy or early childhood phenotype-genotype heterogeneity exists in newborns, phenotypes are fatal and often go unrecognized (many symptoms, can be misdiagnosed) milder symptoms may be undiagnosed for years CNS involvement (neuro disorders, mental disorders) organomegaly (spleen, liver) connective tissue ocular pathology (something wrong with eye nerves) |
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
LSD causes
|
primary (genetic) or secondary (changes cellular processes, secondary biochemical pathways)
|
|
|
|
glycogen storage disease 2
(Pompe) |
LSD
|
|
|
|
sphingolipidoses
|
Fabry, Farber, Gaucher, Tay-Sachs, Niemann-Pick, GM2 Gangliosidosis Sandhoff, Krabbe, MLD
|
|
|
|
Mucopolysaccharidoses
|
MPSI or Hurler, MPSII or Hunter, MPSIII or Sanfilippo, MPSIV or MOrquio, MPSVI or Maroteaux-Lamy, MPSVII or SLy
|
|
|
|
other LSDs
|
glycoproteinoses, neuronal ceroid liposuscinoses, transport defects
|
|
|
|
mucolipidoses
|
MLII, MLIII
|
|
|
|
treatment of LSDs
|
based on the disease pathway, enzyme to substrate (low vs high) defective lysosomal enzyme is a hydrolase
-protein re-engineering -bone marrow and stem cell transplantation -gene therapy -enzyme replacement therapy (ERT) enzyme enhancement therapy (chaperones) substrate reduction therapy identify secondary biochemical pathways affected by substrate accumulation and try to intervene. |
|
|
|
Pompe (GSD II)
|
due to a missing lysosomal debranching enzyme in lysosome, alpha-glucosidase
results in accumulation of glycogen (cant break down to glucose) because in lysosome, disease doesnt contribute to hypoglycemia contributes to skeletal and cardiac muscles and liver lysosomes normally small, become distended three forms -infantile death by two years progressive muscle weakness profound hypotonia macroglossia (enlarged tongue) cardiomegaly and cardiac failure -Late Juve/Adult form milder than infantile, greater variation in symptom onset age some glucosidase activty (some debranching enzyme present) progressive proximal weakness in trunk exercise intolerance hypotonia hepatomegaly absence of severe cardiac involvement (some slight) respiratory insufficiency death usually respiratory failure |
|
|
|
sphingolipidoses
|
LSD where enzyme affected is generally lysosomal hydrolase (degrades sphingolipids)
deficiency of enzyme itself or lack of an enzyme transporter in lysosomal membrane accumulation of sphingolipids hallmark symptom is progressive neurodegeneration autosomal recessive disorder (Fabry's is X linked) MR is common to many of these diseases sphingolipids glycosphingolipid is a type of sphingolipid -sphingosine backbone with either a glucose or galactose attached four major classes -cerebrosides (single glucose/galactose moeity) glucocerebroside/galactocerebroside -Globosides similar to cerebroside, but has 2+ sugar molecules -Gangliosides similar to globosides, but has a sialic acid -Sulfatides have sulfuric acid attached by an ester to galactocerebroside sphingosine+FA--> ceramide phosphate+alcohol--> sphingomyelin glycosphingolipid must have glucose or galactose globosides have more than one sugar galgliosides have sialic acid ceramide sphingolipids plus FA create ceramide (add more sugar, get cerebrosides (gluco/galactocerebrosides) degraded by enzymes -galactocylceramidase (krabbe disorder if missing) -glucocylceramidase (gaucher disorder if missing) Hexosaminidase A or B def. is Tay-Sachs or Sandoff's Alpha-galactosidase A def. results in Fabry's |
|
|
|
Tay Sachs
|
def. Hexasoaminidase A which catalyzes degradation of gangliosides GM2
gangliosides are synthesized and degraded very rapidly (important in early life and brain development) accumulation of gangliosides in tissues is harmful and can be fatal development is normal for first few months, then symptoms appear -nerve cells become distended with fatty materials -affects mental function and physical abilties -Lose all muscle tone carriers of disease are missing enzyme presently no treatment (anticonvulsants for seizure control) Ashkenazi Jews mostly Affects symptoms (cherry red spots in eyes, blindness, deafness, inability to swallow due to muscle atrophy and paralysis dementia, seizures, late age of onset can be 20-30 years (very rare) normal sounds abruptly startle child 1/27 jews are carriers, 1/3000 jewish babies born with disease |
|
|
|
Fabry's
|
deficiency of alpha-galactosidase enzyme
mutation causes buildup of globosides or ceramide trihexoside (accumulates in eyes, kidneys, nervous tissue, CV system) X linked lipid storage disease enzyme replacement therapy approved by FDA is used for Tx patients die prematurely from strokes, heart disease, renal failure symptoms -burning sensation when sweat or exercise corneal cloudiness in eye angiokeratomas (raised reddish blemishes on skin of chest and back) renal failure |
|
|
|
Metachromatic Leukodystrophy
|
MLD
lack of aryl sulfutase A, results in buildup of sulfatides lipid is important for protection on myelin sheath and what surrounds nerve tissue kidneys,gallbladder, other organs affects autosomal recessive three forms late infantile begins 1-2 years old juvenile begins 4-12 years old adult/late stage juvenile after 14 years symptoms issues with muscle tone and movement walking, ,muscle control, falls, feeding, swallowing difficulties decreased mental function behavior issues nerve functions in general metachromatic granules |
|
|
|
gaucher disease
|
defect in beta-glucosidase
accumulation of glucocerebroside -accumulates in macrophages, histologically called Gaucher cells can causes splenomegaly and hepatomegaly, issues with bone marrow and anemia three types type I most common in general and ashkenazi jewish bone disease, anemia, splenomegaly type II disease infancy, neurological involvement, rapidly lead to early death type III spleen, liver, brain problems patient has longer life MR loss of bone density, fractures, bone pain seizures |
|
|
|
gaucher disease
|
defect in beta-glucosidase
accumulation of glucocerebroside -accumulates in macrophages, histologically called Gaucher cells can causes splenomegaly and hepatomegaly, issues with bone marrow and anemia three types type I most common in general and ashkenazi jewish bone disease, anemia, splenomegaly type II disease infancy, neurological involvement, rapidly lead to early death type III spleen, liver, brain problems patient has longer life MR loss of bone density, fractures, bone pain seizures |
|
|
|
Neimann Pick disease
|
accumulation of sphingomyelin in liver, spleen, lung, bone marrow, brain
multi organ involvement causes many different symptoms enlargement of organs classic type A variant is a missense mutation that leads to complete deficiency of the sphingomyelinase enzyme affected cells become enlarged histologically lipid laden macrophages (sea-blue histiocytes) age of onset is different Type I Type A (first few months after birth) abdominal area gets larger cherry red spots in eye loss of motor skills early in life type B milder, occurs later in childhood, adolescence abdominal swelling, splenomegaly no brain or nervous system involvement no loss of motor skills Type II type C (handout) Type D similar to type c |
|
|
|
Neimann Pick disease
|
accumulation of sphingomyelin in liver, spleen, lung, bone marrow, brain
multi organ involvement causes many different symptoms enlargement of organs classic type A variant is a missense mutation that leads to complete deficiency of the sphingomyelinase enzyme affected cells become enlarged histologically lipid laden macrophages (sea-blue histiocytes) age of onset is different Type I Type A (first few months after birth) abdominal area gets larger cherry red spots in eye loss of motor skills early in life type B milder, occurs later in childhood, adolescence abdominal swelling, splenomegaly no brain or nervous system involvement no loss of motor skills Type II type C (handout) Type D similar to type c |
|
|
|
Sandhoff disease, GM1 gangliosidosis, Krabbe
|
handout
|
|
|
|
Sandhoff disease, GM1 gangliosidosis, Krabbe
|
handout
|
|
|
|
Mucopolysaccharidoses MPS
|
MPS I Hurler and Scheie syndromes
hurler-most severe developmental delays, severe respiratory, do not live beyond ten scheie: less severe normal intelligence, less progressive physical problems, normal looking features can live to decades of age MPS II Hunter Disease MPS III-IX normal degradation of carbohydrate chains of glycosaminoglycans defective enzyme that is important in the glycosaminoglycan pathway can be normal intellect to profound MR symptoms rough facial features (gargoyle-like), thick lips, enlarged mouth and tongue dwarfism, abnormal bone size/shape thickened skin hepato/splenomegaly mucopolysaccharides (glycosaminoglycans) are involved in MPS glucosamine glycans derived from glucose heparin and dermatan sulfate are derived from glucose -component of joints, synovial fluid, lubricants, collagen binders, anticoagulant (heparin) hyaluronic acid -improves viscosity and elasticity of synovial fluid tx for osteoarthrities corrects facial wrinkles/folds MPS I: Hurler def. alpha-L-iduronidase accumulation of dermatan and heparin sulfate causes tissue and organ dysfunction broad mouth, square jaw, receding chin corneal clouding dwarfism bc joint deformities MPS II HUnter's syndrome def. enzyme iduronate sulfatase still accumulate dermatan/heparin sulfate Xlinked no corneal clouding |
|
|
|
Mucopolysaccharidoses MPS
|
MPS I Hurler and Scheie syndromes
hurler-most severe developmental delays, severe respiratory, do not live beyond ten scheie: less severe normal intelligence, less progressive physical problems, normal looking features can live to decades of age MPS II Hunter Disease MPS III-IX normal degradation of carbohydrate chains of glycosaminoglycans defective enzyme that is important in the glycosaminoglycan pathway can be normal intellect to profound MR symptoms rough facial features (gargoyle-like), thick lips, enlarged mouth and tongue dwarfism, abnormal bone size/shape thickened skin hepato/splenomegaly mucopolysaccharides (glycosaminoglycans) are involved in MPS glucosamine glycans derived from glucose heparin and dermatan sulfate are derived from glucose -component of joints, synovial fluid, lubricants, collagen binders, anticoagulant (heparin) hyaluronic acid -improves viscosity and elasticity of synovial fluid tx for osteoarthrities corrects facial wrinkles/folds MPS I: Hurler def. alpha-L-iduronidase accumulation of dermatan and heparin sulfate causes tissue and organ dysfunction broad mouth, square jaw, receding chin corneal clouding dwarfism bc joint deformities MPS II HUnter's syndrome def. enzyme iduronate sulfatase still accumulate dermatan/heparin sulfate Xlinked no corneal clouding |
|
|
|
Mucolipidoses
|
handout
|
|
|
|
Mucolipidoses
|
handout
|
|
|
|
ways to measure metals
|
chemical
-chemical chelator (will bind and change color, spectrophotometric assay) flame inductive analysis -burn, observe light refractions (more accurate, more sensitive) ICP-MS (inductively coupled plasma mass spectrometry) burn then run through mass spec picks up many contaminants difficult to discern between mineral and contaminant |
|
|
|
Mg and Ca
|
soft alkaline metals
diffuse in and out because not tightly bound to enzymes concentration is important |
|
|
|
transition metals
|
tend to be held more tightly to proteins
bound for functionality (co-factors) but also safety, dont want them freely floating around body some are not necessary but may be helpful (fluoride stabilize tooth enamel) |
|
|
|
water and salts
|
intra/extracellular ion concentration can differe by 2 orders of magnitude
gradient drives cellular reactions (i.e. symporter diffusion H+ pulls in lactose) (i.e. symporter pulls glucose into cell on one side, falls down gradient in other) cell membranes permeable to H2) (aquaporins and ion channels allow this) need to maintain solute/water concentration to prevent lysis or cell shrinking extracellular fluids act as conduit around cells, using entire surface area for exchange volume estimate for 73kg person 24L intracellular 16L liquid extracelular -interstitial volume 11.2L plasma volume 3.2 L Transcellular (inside organs) 1.6L TBW estimate (Liters) = BW(lbs)/4 subtract 10% for obese person, add 10% for lean person women have 10% less water than man of similar bodyweight osmolality is particle(colute) concentration of fluid, units millosmoles/kg (mos-mol/kg) Eextracellular fluid osmolality=intracellular fluid (to prevent lysis/shrinking) ECF solutes (or osmoles) are very different from ICF due to action of transporters and active pumps and diff. permeability of membrances ECF solutes are Na, Cl, HCO3 ICF solutes are K and organic phosphate esters (ATP, creatine phosphate, phospholipids, UTP, GTP,) contributes to electrical charge some soutes restricted to either ecf or icf and determine the effective osmolality of that compartment since Na is mostly ECF, total body content reflects the ECF, same for K and ICF cells can alter these levels in extreme situations proteins estimated to be 15mEq, but difficult to measure due to size differences serum albumin responsible for ~80% osmotic pressure of blood milliequivalents=millimolar (when only one ionizable group) mEq=2XmMolar if there are two charges mmol stays the same, must adjust for equivalents if there are multiple ionizable groups with different pKs it is more pH dependent proteins average to about 15mEw because they have many ionizable charges dependent on pH and salt concentrations |
|
|
|
water and sodium
|
Na and K are main macrominerals
normal osmolality of plasma is 275-290 mosmol/kg and maintained by mechanisms that can detect 1-2% changes avg western diet exceeds 150mmoles NACL per day (more than required) avg 70kg man contains 100g sodium, half in bone, 40% ecf recommended intake 2400, usual intake 3000-6000mg/day requirement 500mg/day can lead to hypertension, is a genetic component to na sensitivity and concentrations will affect people differently |
|
|
|
water and sodium loss
|
water is lost from skin by sweating, liquid gets on surface, evaporates away, increases entropy of system, takes away energy, cools you off
mainly depends on heat and caloric expenditure water is lost by exhaling, but also same amount created by metabolism (end product of cytochrome oxidase in ETC) net activity of GI tract down to jejunum is secretion of water and electrolytes net activty from jejunum to colon is reabsorption of water typically ~100mL water per day excreted with feces ingested food/water becomes isotonic so if vomit or have diarrhea lose ions (cholera, lose water and ions, die quickly) diarrheal fluid is close to isotonic, can lose liters water/salt loss in urine is variable |
|
|
|
water and potassium
|
recommended intake 3500mg
usual 4000-5000/day requirement 2000mg too much K will disrupt normal cellular function and cause death na/k ratio linked to hypertension deficiency of K heart arrythmias, muscle weakness, increased blood pH (alkalosis) excess K cardiac arrest, |
|
|
|
Calcium
|
regulate intracellular enzyme activities
secretory processes (nerve conductance, pancreatic enzymes, milk protein release) blood clotting (K assists in binding gamma-carboxy-gluatamate to Ca) muscle contraction structure/growth of bones and teeth binds to proteins affecting function intracellular Ca can be .1uM (10,000x lower than ECF) large variation in magnitude allows for dynamic range in cellular signaling second messenger function hormone-receptor interactions -i.e. epi released from adrenal medulla, binds alpha receptors in liver, activates glycogenolysis and inhibits glycogen synthesis mainly by raising Ca levels in liver calmodulin binding regulates many proteins and processes such as muscle contraction and inflammation, is a key regulator of many Ca dependent proceses Ca and Vitamin D are intimately linked together in terms of uptake and function in the body absorption of Ca affected by Vit D gastric Acid (makes more soluble) lactose citrate, malate, protein, phosphorous exercise inhibited by oxalic acid, phytic acid, dietary fiber phosphate (binds to metals, makes insoluble) steatorrhea (soaps) too much fat getting past small intestine and sweeping stuff out |
|
|
|
calcium deficiencies
|
rickets, osteomalacia (adult rickets) both reversible with supplements
vitamin D prevents ricekts (poor intestinal absorption/poor kidney reabsorption of Ca and Phopshate osteoporosis (problems taking up and mobilizing Ca) adult males should consume 1000mg/day, adolescents and women needmore excessive intake increases risk of renal stone formation (genetic component) |
|
|
|
Phosphorous P (phosphate PO4)
|
second most abundant mineral in body
85% in bones and teeth, 15% elsewhere (including nucleic acids) also regulated by vitamin D functions in structure of nucleic acids, phospholipids, activation of enzymes by phosphorylation, energy ATP also acid-base balance (has many ionizable groups) dietary sources animal protein, milk, eggs processed foods food additive for pH adjustment, to sequester some things (like metals) prevents bacterial growth recommended intake 700-1250mg/day dietary excess is commong, promotes Ca excretion |
|
|
|
Magnesium
|
bone strength
ATP hydrolysis (stabilizes 3- charge of phosphate) most bases floating around cells have an Mg on them Enzyme cofactors (also for structural reasons) almost every enzyme in DNA/RNA reactions require Mg binds nucleic acids directly and changes their shape muscle relaxation after contractions food sources vegetables, nuts, legumes 30-50% of intake is absorbed Intracellular>extracellular deficiency rate except with alcoholics (DTs, hallucinations) hypertension, vascular disease, preeclampsia, osteoporosis excess: anesthetic effects and diarrhea |
|
|
|
Sulfur S (sulfate SO4)
|
sulfate in tissues and sulfur containing amino acids methionine and cysteine
key role in protein structure (Cys-S-S-Cys) disulfide bonds involved in post-translational modification key role in transfer gorups (acetyl CoA) things are easily attached to and removed from S also critical in glutathione (SH a powerful tool) no issue with def./excess as long as have normal diet |
|
|
|
iron
|
key role in many enzymes
ETC -complexes 1,2,3,4, iron in form of single iron atoms, iron-sulfur center that spontaneously form, iron sitting in heme and cytochromes can transfer electrons can bind oxygen can catalyze reactions and do many things depending on its surroundings transition metals, especially iron, are the molecular equivalent of nuclear fuel iron and manganese are critical to body, but can be poisonous can react with oxygen and make superoxide reduced form can interact with hydrogen peroxide and make a hydroxyl radical (free radical with unpaired electron) can destroy anything they touch some enzymes make these on purpose these metals can also be used to protect catalase-destroys hydrogen peroxide (has heme in it) Fe is redox active can transfer electrons either way Fe2+/Fe3+ reduction potential is the relative ability to give or accept electrons the more negative, the more likely it will give up electrons, the more positive the more likely it will accept redox midpoint potential, way to estimate an enzyme's ability to bind a substrate electrical potential at which molecule is 50% oxidized and 50% reduced cells use transition metals because they can be easily moved from one oxidation state to another without giving dangerous free radicals (due to d orbitals) cant do this with organic molecules Cytochrome A,C,B1 all contain iron dramatic difference in reduction potential Cytochrome F (.365mv) vs ferridoxin (-.432mv) key point: proteins can grab a metal and by controlling surrounding organic elements (for example, embedded in heme prosthetic group or iron-sulfur center with different amino acids) they can tune it to whatever potential they want with same metal can have iron at multiple potentials by controlling AAs around it this is how ETC works Flavins are organic rings that take up and give electrons same flavin can have different potentials depending on what proteins are around them main function of iron is redox chemistry average 3-4g iron in body (half in blood via iron atom in hemoglobin) stored as ferritin or hemosiderin ferritin is a huge globe of many different monomers hollow shell that contains blocks of oxidized iron sequestering away from oxygen so that it doesnt react with oxygen and form reactive oxygen species hemosiderin mostly in liver, doles it out as necessary depending on blood concentration males store about a gram, females less iron overload 40-50g, mostly liver, spleen, bone marrow myoglobin in muscle stores iron ETC enzymes only 8mg to run enzymatic machines in cells (most is stored or used to transport oxygen) strictly regulated iron metabolism Fe2+ is normal form, Fe3is very insoluble reduction is activated by vitamin C iron reaches plasma and is carried by transferrin cells that need iron put a transferring protein receptor on surface cells pick it up and store it as ferritin iron can be stored in bone every cell needs it RBC huge amounts reticular endothelial sells use it well loss liver sweeps iron into feces with bile salts sweat losing skin bleeding intestinal absorption divalent metal transporter (DMT) -transports many different metals enzymes are blind that feel for certain structure metals have similar overall shape, so they arent really differentiated by transporter much is unknown about metal uptake in cells taken up in 2+ form, also taken up as heme and citrate once in intestinal cells, most will be stored as ferritin ultimately shipped to blood stream on transferrin iron at least 8mg per day to maintain stores since we lose about 8mg per day RDAs higher when developing females need more after menstruation ends and more during pregnancy for fetal development genetically may be individual needs, we are unaware iron uptake by transferrin when cells have transferrin receptor on the surface, they bind the transferrin, then release iron by acidification in lysosomes iron is then grabbed by internal proteins for storage some metals are soluble in high acid, so easier to access in acidic condition non-heme iron uptake diet affects uptake organic acids from plants, phytic acids polythenols -i.e. resveratrol -have lots of OH that can grab metals, coordinate them and sweep them up in your diet black tea and cocoa bind iron tightly drink lots of black tea, wash iron from diet can stimualte uptake Vitamin C organic acids (i.e. citric acid) heme |
|
|
|
iron excess
|
hereditary defects
hemochromatosis despite rarity, will see these in testing iron overload dietary overload is hemosiderosis fad diet or heavy consumptoon of red wine or overdose of iron supplements huge amount of oxidative stress extra iron reacts with oxygen in body some tissues are more sensitive (cardiac issues) frataxin protein controls Fe levels in mitochondria friedrich's ataxia-defective frataxin, buildup of iron in mitochondria excess also decreases absorption of hormones and antibiotics moderately high levels might be bad, body will try and maintain concentration at vigorous levels infecting bacterium has no available iron some bacterium cause hemolysis to release iron because they need it |
|
|
|
iron deficiency
|
1 billion anemic people
dietary deficiencies infection, heliobacter ulcers vitamin deficiencies since DMTs are non discriminatory, you may get lead poisoning transporter is waiting for iron,but grabbing other metals because similar in structure inflammation malabsorption first thing seen in deificiency is anemia |
|
|
|
Zinc
|
roles in many enzymes
involved with proteins that interact with DNA (zinc fingers) histidines or cysteines that grab on to zinc dna is a negative nucleic acid and zinc is positive may have been evolutionary chosen zinc over iron because not redox active (cant add and take away electrons in biological conditions) shaped and charged like iron but doesnt cause reactive oxygen species hard to measure because it many things,hard to discriminate from background noise uptake/absorption histidines, cysteines, and phytic acid bind zinc tightly and sweep into diet metallithioneins proteins with 10-12 cysteines regulation of movement and storage of zine through cells |
|
|
|
intestinal absorption of zinc
|
much is unknown
once Zn is in, metallothioneins bind to them might be specific transporters in blood, serum albumin may carry it as well |
|
|
|
zinc storage
|
couple of grams, women less than men
turnover takes about a year, but estimated 1mg lost per day (skin loss) liver turnover is faster homeostasis regulates Zn levels in a tight range |
|
|
|
zinc deficiency
|
take about 10mg/day
many enzymes require zinc tissue damage, increase oxidative stress, immune deficiencies, developmental changes, skin lesions, children develop later, not enough zinc for regulatory proteins that control cellular development later onset of secondary sex characteristics poor appetite (zinc linked to taste) malabsorption diseases affect all minerals (i.e. IBS) |
|
|
|
zinc excess
|
common belief zinc is a cure all
excess: nausea (50mg can have emetic effect) because of DMT, too much of any mineral can affect others (ie iron and copper) implicated in alzheimer's disease can lower HDLs and increase cholesterols in bloodstream |
|
|
|
copper
|
critical for complex 4- terminal oxygen reducer in ETC
has two ox states, 1+ and 2+ reduced copper is insoluble implicated in prions and mad cow disease (bovine and human spongiform encephalopathy) actual protein binds copper deficiency can lead to anemia copper uptake is linked to Fe uptake if copper is deficient, Fe uptake is also deficient, leading to anemia Deficiency: anemia, leukopenia, neutropenia, osteoporosis, seen in total parenteral nutrition (TPN) excess can also cause anemia, indirectly. if you limit copper uptake, you limit iron uptake excess: weakness, tremors genetic diseases wilson's disease (like hemochromatosis) lvier cant get rid of copper, there is a buildup early brain damage from ixidative stress, involuntary movements, psychoses, general liver damage, cirrhosis classic greenish-gold ring in cornea from copper buildup liver transplant or copper binding drugs |
|
|
|
iodine
|
not really metal
critical to thyroid hormone - growth development, metabolism micrograms per day concentrated in thyroid covalently bound to tyrosines in proteins regulates metabolic rates deficiencies: spontaneous abortion, irreversible brain damage symptom in adult (goiter, enlarged thyroid) excesses, no apparent effects, but can disrupt thyroid function like deficiency at chronic excess most table salts have iodine |
|
|
|
selenium
|
not an antioxidant, but involved in antioxidant processes
critical component of posttranslation modification of proteins (selenomethionine and selenocysteine) similar electron shell to sulfur, but selenium is different enough to use it instead of sulfur for certain purposes (usually with proteins that have antioxidant defense) excess: deposits into all sulfur proteins, when not every protein can use selenium in place of sulfur adults require 55ug/day deficiencies -cardiac failure (sensitive to oxidative stress bc many mitochondria) liver problems cancer atherosclerosis hair loss (skin changes) dermatitis seen in both excess and deficiency peripheral neuropathy in excess uptake is not well understood, hard to study because so little of it |
|
|
|
synthesis, processing, secretion VLDL
|
Apo B100 protein (attached to lipoprotein) are synthesized on RER
Triacylglycerol (TGs) are synthesized on SER and/or on FA synthase in cytosol TG and proteins are packaged in the Golgi complex to form VLDL VLDL are transported to cell membrane in secretory vesicles and secreted by exocytosis cholesterol located near surface because has OH group and is amphipathic |
|
|
|
Apo B100 and Apo B48
|
made from same genes
Apo B100 found in VLDL and synthesized in liver, twice as long as ApoB48 Apo B48 found in chylomicrons and synthesized in intestinal cells Different transcripts due to RNA editing Apo B48 has UAA instead of CAA (introduces stop codon in intestinal cells) VLDL major component is TG (not as much as chylomicrons) epithelial cells lining gut will be packaging up the chylomicrons go out into bloodstream find an enzyme on lumen side of blood vessel called lipoprotein lipase LPL takes off FA from VLDL, FA go into adipose and packaged as TGs and stored Glucose comes in to make backbone of VLDL, Glycerol-3-P, which combines with FA CoA to make TG, which are then packaged as VLDL VLDL meets LPL, breaks down TG to FA and glycerol and goes into adipose, becomes again TG LPL works on chylomicrons and VLDL (both have pool TGs) LPL is dependent on insulin adipose only has one path to Glycerol-3-Phosphate via glucose to prevent futile cycle of TG synthesis and breakdown insulin stimulates transport of glucose into adipose cells and stimulates synthesis and secretion of LPL ApoCII obtained from HDL activates LPL (present on both VLDL and chylomicrons) FA to TG in liver, sent into blood as VLDL Acetyl CoA carboxylase regulated enzyme |
|
|
|
total cholesterol (TC)
|
should be less than 200 mg/dL
formula is TC=HDL + LDL + (TG/5) |
|
|
|
TG
|
refers to chylomicrons and VLDL on tests
|
|
|
|
chylomicrons
|
dietary, produced by cells lining small intestine
80% TG |
|
|
|
VLDL
|
synthesized in liver
increase TG approximately 45% TG |
|
|
|
LDL
|
carriers cholesterol to tissue
a lot of cholesterol |
|
|
|
HDL
|
reverse transports cholesterol to liver
composed of more proteins compared to other constituents |
|
|
|
lipoproteins and transport
|
Lipids (TG, FA)
chylomicrons transport from intestines to tissues VLDL transports from liver to tissues adipose to muscle and liver by FA on albumin in blood cholesterol to tissues by LDL from tissues by HDL |
|
|
|
structure of lipoprotein
|
apoproteins (binding things together, some towards center some out to liquid environment
amphipathic liquids both charged and non charged parts phospholipids have charge some come in contact with liquid environment some extends into center that is less charged cholesterol - OH group not esterified. doesnt have an FA attached to it non polar lipids located on the inside of the phospholipid cholesterol esters - attached to LCFA TG=no charge Lipoproteins are different sizes chylomicrons are huge then VLDL then LDL then HDL |
|
|
|
Apo A1
|
associated with HDL
activates LCAT/PCAT (same thing) in order to take cholesterol to cholesterol ester ligand for HDL receptor chylomicrons |
|
|
|
ApoB100
|
LDL, VLDL, IDL
LDL receptor ligand |
|
|
|
ApoB48
|
chylomicrons, chylo remnants,
chylo assembly and secretion, dietary lipids |
|
|
|
ApoCI
|
VLDL, HDL, Chylomicrons
may activate LCAT may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
APO CII
|
VLDL, HDL, Chylomicrons
Cofactor, activates LPL, is transferred between HDL and VLDL/chylomicrons |
|
|
|
ApoCIII
|
VLDL, HDL, CHylomicrons
|
|
|
|
ApoE
|
VLDL, HDL, CHylomicrons, chylo remnants, LDL
hepatic receptor ligands for chylomicrons and LDL is recycled between HDL and VLDL/chylomicrons |
|
|
|
both ApoB100 and ApoB48 encoded by same gene
|
apoB100 attaches to LDL receptor
Deamination step whic changes the RNA by RNA editing from a C to a U changing CAA (gln) to UAA (stop) ApoB48 codes first half of transcript liver cells do not synthesize deaminase, so the resulting transcript and protein are longer than in intestinal cells. Apo B100 is about twice as long as ApoB48 and will attach to LDL receptor ApoB48 will not |
|
|
|
Apo A1
|
associated with HDL
activates LCAT/PCAT (same thing) in order to take cholesterol to cholesterol ester ligand for HDL receptor chylomicrons |
|
|
|
ApoB100
|
LDL, VLDL, IDL
LDL receptor ligand |
|
|
|
ApoB48
|
chylomicrons, chylo remnants,
chylo assembly and secretion, dietary lipids |
|
|
|
ApoCI
|
VLDL, HDL, Chylomicrons
may activate LCAT may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
APO CII
|
VLDL, HDL, Chylomicrons
Cofactor, activates LPL, is transferred between HDL and VLDL/chylomicrons |
|
|
|
ApoCIII
|
VLDL, HDL, CHylomicrons
|
|
|
|
ApoE
|
VLDL, HDL, CHylomicrons, chylo remnants, LDL
hepatic receptor ligands for chylomicrons and LDL is recycled between HDL and VLDL/chylomicrons |
|
|
|
both ApoB100 and ApoB48 encoded by same gene
|
apoB100 attaches to LDL receptor
Deamination step whic changes the RNA by RNA editing from a C to a U changing CAA (gln) to UAA (stop) ApoB48 codes first half of transcript liver cells do not synthesize deaminase, so the resulting transcript and protein are longer than in intestinal cells. Apo B100 is about twice as long as ApoB48 and will attach to LDL receptor ApoB48 will not |
|
|
|
synthesis, processing, secretion VLDL
|
Apo B100 protein (attached to lipoprotein) are synthesized on RER
Triacylglycerol (TGs) are synthesized on SER and/or on FA synthase in cytosol TG and proteins are packaged in the Golgi complex to form VLDL VLDL are transported to cell membrane in secretory vesicles and secreted by exocytosis cholesterol located near surface because has OH group and is amphipathic |
|
|
|
Apo B100 and Apo B48
|
made from same genes
Apo B100 found in VLDL and synthesized in liver, twice as long as ApoB48 Apo B48 found in chylomicrons and synthesized in intestinal cells Different transcripts due to RNA editing Apo B48 has UAA instead of CAA (introduces stop codon in intestinal cells) VLDL major component is TG (not as much as chylomicrons) epithelial cells lining gut will be packaging up the chylomicrons go out into bloodstream find an enzyme on lumen side of blood vessel called lipoprotein lipase LPL takes off FA from VLDL, FA go into adipose and packaged as TGs and stored Glucose comes in to make backbone of VLDL, Glycerol-3-P, which combines with FA CoA to make TG, which are then packaged as VLDL VLDL meets LPL, breaks down TG to FA and glycerol and goes into adipose, becomes again TG LPL works on chylomicrons and VLDL (both have pool TGs) LPL is dependent on insulin adipose only has one path to Glycerol-3-Phosphate via glucose to prevent futile cycle of TG synthesis and breakdown insulin stimulates transport of glucose into adipose cells and stimulates synthesis and secretion of LPL ApoCII obtained from HDL activates LPL (present on both VLDL and chylomicrons) FA to TG in liver, sent into blood as VLDL Acetyl CoA carboxylase regulated enzyme |
|
|
|
total cholesterol (TC)
|
should be less than 200 mg/dL
formula is TC=HDL + LDL + (TG/5) |
|
|
|
TG
|
refers to chylomicrons and VLDL on tests
|
|
|
|
chylomicrons
|
dietary, produced by cells lining small intestine
80% TG |
|
|
|
VLDL
|
synthesized in liver
increase TG approximately 45% TG |
|
|
|
LDL
|
carriers cholesterol to tissue
a lot of cholesterol |
|
|
|
HDL
|
reverse transports cholesterol to liver
composed of more proteins compared to other constituents |
|
|
|
lipoproteins and transport
|
Lipids (TG, FA)
chylomicrons transport from intestines to tissues VLDL transports from liver to tissues adipose to muscle and liver by FA on albumin in blood cholesterol to tissues by LDL from tissues by HDL |
|
|
|
structure of lipoprotein
|
apoproteins (binding things together, some towards center some out to liquid environment
amphipathic liquids both charged and non charged parts phospholipids have charge some come in contact with liquid environment some extends into center that is less charged cholesterol - OH group not esterified. doesnt have an FA attached to it non polar lipids located on the inside of the phospholipid cholesterol esters - attached to LCFA TG=no charge Lipoproteins are different sizes chylomicrons are huge then VLDL then LDL then HDL |
|
|
|
Apo A1
|
associated with HDL
activates LCAT/PCAT (same thing) in order to take cholesterol to cholesterol ester ligand for HDL receptor chylomicrons |
|
|
|
Apo A1
|
associated with HDL
activates LCAT/PCAT (same thing) in order to take cholesterol to cholesterol ester ligand for HDL receptor chylomicrons |
|
|
|
ApoB100
|
LDL, VLDL, IDL
LDL receptor ligand |
|
|
|
ApoB100
|
LDL, VLDL, IDL
LDL receptor ligand |
|
|
|
ApoB48
|
chylomicrons, chylo remnants,
chylo assembly and secretion, dietary lipids |
|
|
|
ApoB48
|
chylomicrons, chylo remnants,
chylo assembly and secretion, dietary lipids |
|
|
|
ApoCI
|
VLDL, HDL, Chylomicrons
may activate LCAT may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
ApoCI
|
VLDL, HDL, Chylomicrons
may activate LCAT may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
APO CII
|
VLDL, HDL, Chylomicrons
Cofactor, activates LPL, is transferred between HDL and VLDL/chylomicrons |
|
|
|
APO CII
|
VLDL, HDL, Chylomicrons
Cofactor, activates LPL, is transferred between HDL and VLDL/chylomicrons |
|
|
|
ApoCIII
|
VLDL, HDL, CHylomicrons
inhibits LPL, may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
ApoCIII
|
VLDL, HDL, CHylomicrons
inhibits LPL, may inhibit hepatic uptake of chylo and VLDL remnants |
|
|
|
ApoE
|
VLDL, HDL, CHylomicrons, chylo remnants, LDL
hepatic receptor ligands for chylomicrons and LDL is recycled between HDL and VLDL/chylomicrons |
|
|
|
ApoE
|
VLDL, HDL, CHylomicrons, chylo remnants, LDL
hepatic receptor ligands for chylomicrons and LDL is recycled between HDL and VLDL/chylomicrons |
|
|
|
both ApoB100 and ApoB48 encoded by same gene
|
apoB100 attaches to LDL receptor
Deamination step whic changes the RNA by RNA editing from a C to a U changing CAA (gln) to UAA (stop) ApoB48 codes first half of transcript liver cells do not synthesize deaminase, so the resulting transcript and protein are longer than in intestinal cells. Apo B100 is about twice as long as ApoB48 and will attach to LDL receptor ApoB48 will not |
|
|
|
Pathway for chylomicrons (CM)
|
small intestinal cell made nascent chylomicron with ApoB48 attached
composed of more TG than cholesterol out in bloodstream ApoCII and ApoE are transferred from HDL to nascent CM CM goes through capillaries and encounters LPL which takes TGs down to FAs and glycerol extracellular LPL activated by ApoCII degrades TG in CM and VLDL to FAs which are taken up by adipose and resynthesized to TGs as chylomicrons lose TGs they decrease size and become chylo remnants CM remnants have more cholesterol esters than TGs CM remnants no longer need ApoCII and give back to HDL Keeps ApoE because it binds to receptor in liver which allows it to be taken up cholesterol from CM may be used in liver to make bile or in membranes if liver has enough cholesterol it will repackage as VLDL and send it back out LDL binds to specific receptors on extrahepatic tissues and on the liver where they are endocytosed |
|
|
|
both ApoB100 and ApoB48 encoded by same gene
|
apoB100 attaches to LDL receptor
Deamination step whic changes the RNA by RNA editing from a C to a U changing CAA (gln) to UAA (stop) ApoB48 codes first half of transcript liver cells do not synthesize deaminase, so the resulting transcript and protein are longer than in intestinal cells. Apo B100 is about twice as long as ApoB48 and will attach to LDL receptor ApoB48 will not |
|
|
|
Pathway for chylomicrons (CM)
|
small intestinal cell made nascent chylomicron with ApoB48 attached
composed of more TG than cholesterol out in bloodstream ApoCII and ApoE are transferred from HDL to nascent CM CM goes through capillaries and encounters LPL which takes TGs down to FAs and glycerol extracellular LPL activated by ApoCII degrades TG in CM and VLDL to FAs which are taken up by adipose and resynthesized to TGs as chylomicrons lose TGs they decrease size and become chylo remnants CM remnants have more cholesterol esters than TGs CM remnants no longer need ApoCII and give back to HDL Keeps ApoE because it binds to receptor in liver which allows it to be taken up cholesterol from CM may be used in liver to make bile or in membranes if liver has enough cholesterol it will repackage as VLDL and send it back out LDL binds to specific receptors on extrahepatic tissues and on the liver where they are endocytosed |
|
|
|
pathway of VLDL
|
made in liver with ApoB100
has lots of TGs nascent VLDL goes out into bloodstream HDL gives it ApoCII and ApoE goes through capillaries and encounters LPL and the TGs are broken down into FAs and gylcerol FAs stored in adipose VLDL decreases in size and become IDL and finally LDL LDL gives ApoCII and E to HDL can use ApoB100 which can go to any cell in body because every cell in bdoy had LDL receptor because they all need some cholesterol can also go into liver because liver needs cholesterol tissues making cholesterol based hormones especially need cholesterol |
|
|
|
LPL
|
attach to capillary endothelial cells facing lumen of capillaries
synthesis and secretion stimulated by insulin activated by ApoCII implications for diabetics (Type I w no insulin, Type II isnt working well) insulin wont be there to allow glucose to get into adipose tissue and wont be able to synthesize and bring LPL to surface |
|
|
|
wHSL
|
HSL in the adipose
activated by glucagon stimulating the breakdown of TGs to FAs and gylcerol FAs are needed quickly and will travel on albumin |
|
|
|
Chylmicrons
|
made my epithelial cells
derived from dietary lipids nascent chylomicrons have ApoB48 (then aquire ApoCII and Apo E) become chylo remnants when TG:CHol = 1:1 AoE assists in CMRemnant docking in liver |
|
|
|
VLDL
|
made in liver
derived from liver's synthesis of FA plus any FA from chylo remnants packaged as TG nascent VLDL has ApoB100 and ApoCII and ApoE from HDL Goes to IDL and LDL ApoB100 is major apoprotein LDL does not have ApoCII or ApoE LDL carries cholesterol to peripheral tissues which have specific LDL receptors that recognize ApoB100 LDL receptors transport LDL into peripheral tissue via endocytosis intracellular ACAT (acyl CoA cholesterol acyltransferase) takes cholesterol and adds LCFA to become cholesterol ester allows it to be stored |
|
|
|
LDL uptake by cell receptor mediated endocytosis
|
LDL particle has ApoB100 and alot of cholesterol ester
LDL receptor is being synthesized then sent to outside of cell where it is recognized by ApoB100 incorporated into vacuole internalized pH change so it can be broken down Cholesterol comes out as lipid droplets AA and FA can be used for anything Cholesterol ester is stored to be used Cholesterol regulates its own synthesis via HMG-CoA Red regulates LDL receptor synthesis activates ACAT for storage |
|
|
|
ACAT reaction
|
found in cytosol, creates storage form of cholesterol
cholesterol and fatty acyl CoA catalyzed by ACAT leads to cholesterol ester Fatty Acyl CoA is attached to OH of cholesterol cholesterol ester is nonpolar and can be stored as lipid droplet until needed |
|
|
|
unusual forms of LDL
|
LDL is within circulation, but target is outside of circulation
in order for LDL to get to its receptor, it has to squeeze through and out into the ECM how long it stays in ECM will determine some of the problems associated with Coronary Artery Disease |
|
|
|
Lipoprotein A
|
LDL bound to apolipoprotein A
having too much is a risk factor for CAD ApoA is structurally similar to plasminogen (attaches to blood clots to break them down) but no enzymatic activity because it binds to the clots, it competes with plasminogen and prevents clot breakdown antithrombolytic, procoagulant capacities may not be all bad |
|
|
|
small dense LDL
|
more artherogenic
can squeeze in easier into ECM and has tendency to stay there longer lower binding affinity for LDL receptor high penetration into arterial wall Kringles-binds to clot so plasminogen digests it |
|
|
|
HDL
|
reverse cholesterol transporter
ApoA-1 synthesis in liver and sent into bloodstrea, encounters peripheral tissue complex interactions with ABCA-1 ABCA-1functions -brings cholesterol out to surface so HDl can get it -takes in ApoA-1 (there is series of rxns leading to ApoA1 coming out as nascent HDL) becomes nascent HDL (like flat pancake because phospholipids and cholesterol, but no non polar components to make it round PCAT converts cholesterol to esters deriveing FA from phosphatidylcholine -this enzyme is associated with HDL as make more esters, it will get round there is exchange occuring between VLDL and HDL via action of two enzymes HDL is taking FAs off of phospholipids to make cholesterol esters eventually it runs out of phospholipids, needs to make an exchange for new phospholipids sometimes HDL can go right into liver through hepatic lipase or it can go past liver and give up some cholesterol in the liver and keep going |
|
|
|
HDL
|
synthesized from liver as ApoA1 and secreted into bloodstream
transfers ApoCII and ApoE back and forth from CM and VLDL Drives by ABCA1 to pick up cholesterol and take back to liver called "reverse cholesterol transport) since its returning to liver ABCA1s job is to get cholesterol out of tissue and on to outer leaflet of tissue general functions include antioxidant activity inhibiting platelet activity contains PCAT (phosphatidylcholine cholesterol acyl transferase) enzyme that transforms cholesterol into a non polar cholesterol ester reaction involves taking a FA off of phosphatidylcholine and transferring it to cholesterol reaction is stimulated by apoA1 deficiency with this enzymes results in FishEye disease -HDL not having many esters, roughly 20% of total cholesterol patient presents with opacity in their cornea, which is dense and white, similar to eyes of boiled fish otherwise benign |
|
|
|
ABCA1 loads HDL with cholesterol from tissues
|
ABCA1 is ATP binding cassette 1
part of family of ~20 ABCs function is to transport molecules in and out of tissues one example is drugs, whether it is good or bad depends on what drug is doing in tissues also bile transport loads HDL with cholesterol from tissues |
|
|
|
Tangier Disease
|
patient presents with enlarged, orange yellow tonsils
these people have very low HDL, sometimes barely any as a result, tonsils collect a lot of cholesterol disease state is a result of mutation ABCA1 -rare autosomal recessive disease cannot eliminate cholesterol from tissues because ABCA1 cannot give to HDL also ApoA1 cannot be processed into HDL |
|
|
|
LP exchange
|
CETP-cholesterol ester transfer protein
PLTP phospholipid transfer protein transports molecules between HDL and VLDL HDL can transport phopsholipids to VLDL since it has an abundance of it essentially, enzymes are involved in remodeling of HDL |
|
|
|
Role of liver
|
takes up chylomicron remnants
makes HDL at least the ApoA1 part takes up HDL containing cholesterol on HDLs trip back from tissues synthesizes cholesterol -produces bile using some cholesterol -secretes extra cholesterol into feces synthesizes FAs and uses it to create VLDLs |
|
|
|
Cholesterol comes from Food and Family
|
statin blocks major step in synthesis of cholesterol (HMG-CoA reductase)
|
|
|
|
biosynthesis of cholesterol
|
two enzymes
HMG CoA Synthase makes HMG-CoA (KB synthesis) HMG-CoA synthase can make cholesterol or KBs HMG-CoA reductase second enzyme makes mevalonic acid rate-limiting step, once initiated cholesterol will be synthesized after mevalonic acid synthesis there is a group of isoprenes (groups of 5 carbon structures) enzyme cyclase closes open rings and result is lanosterol after lanosterol, you get cholesterol cholesterol inhibits rate limiting step, HMG-CoA reductase (product inhibition) |
|
|
|
Regulation of HMG CoA reductase
|
first, cholesterol is inhibiting a transcription factor (TF), SCREBP-2
cholesterol binds SCAP -SCAP is attached to SCREBP2, is the precursor form of SCREBP2 binding occurs in ER, sequestering the TF there prevents TF from getting into nucleus and inducing transcription (TS) of HMG-CoA Reductase second event involves degradation of enzyme that makes HMG CoA reductase if sterols are present (if enough cholesterol is present) -under such conditions, enzyme that makes HMG CoA reductase is ubiquinated and targeted for time in proteasome third event is covalent modification by AMP-kinase which phosphorylates HMG-CoA reductase and makes it inactive same type of regulator involved in regulation of acetyl coA carboxylase insulin can use a phosphatase to get that phosphate off (reversing the conditions) and reactivating the enzyme final event involves conditions of either high insulin or thyroxine or low glucagon or low glucocorticoid levels these conditions regulate TS of HMG CoA reductase when insulin or thyroxine is high, that telling cell that it needs HMG CoA reductase around high glucagon or glucocorticoids says no we dont need it so downregulate it |
|
|
|
high cholesterol can sneak up on you
|
shows that liver loads fat onto VLDLs which travel through blood and later become LDL
LDL cholesterol may become stuck as travels through blood, sticking to blood vessel wall HDL take pieces of LDLs back to liver where cholesterol can be repackaged if too much cholesterol in blood, not enough HDL to sequester, can cause blockage and heart attack |
|
|
|
atherosclerosis
|
over time, plaque builds up in blood vessels
eventually, a blood clot has formed and blood vessel is closed off LDL must squeeze through endothelial cells to get to tissues LDLs can be oxidized and stuck in blood vessel walls oxidants can be superoxide, nitric oxide, hydrogen peroxide once oxidized, LDL cannot go back into bloodstream stuck LDLs are phagocytized by monocytes that become macrophages, macrophages take up LDL by two receptors high affinity and non specific receptor become foam cells start accumulating along blood vessel walls contributes to plaque buildup though they are trying to eliminate oxidized LDL lots of calcium=more stiff plaque buildup Vitamin E, ascorbic Acid, Beta-carotene prevent oxidation bilirubin might help as well |
|
|
|
coronary artery disease equivalents
|
precursors that lead to it
-diabetes -aortic aneurysm -peripheral vascular disease -carotid artery disease (stroke, transient ischemic attach) |
|
|
|
framington risk equation
|
age for men is different than women, partially because of menopause
women have higher age before theyre at risk because of estrogen |
|
|
|
metabolic syndrome
|
step 8 of ATPIII
considered to have metabolic syndrome if have three of following abdominal adiposity/obesity high blood pressure hypertriglycerides (VLDLs, LDLs, Chylomicrons, low HDL) Fasting hyperglycemia inflammatory signs (likely due to microalbuminurea caused by albumin excretion through kidneys metabolic syndrome can be reversed to some degree |
|
|
|
vertical auto profile
|
conducted occasionally
can have HDL2 or 3 (2 is more protective) can have LDL A or B (difference not specified) Therefore total amount may not be indicative of good thing |
|
|
|
CHD risk factors
|
similar to ATP III
positive (bad) risk factors include male>45, female>55yrs or premature menopause premature coronary artery disease in close relatives smoking hypertension HDL under 35 mg/dL diabetes obesity HDL above 60mg/dL is negative (good) risk factor) can counteract positive risk factor rsik factors have geometric rise (exponential) |
|
|
|
LDL cholesterol based on risk category
|
LDL under 100 in general
if have low risk, LDL below 160 Moderate risk, LDL below 130 High risk, LDL below 100 beign with therapeutic life changes, dont jump right into drug therapy |
|
|
|
for high risk patients, ATPIIIconsders keeping LDL below 100 a minum goal, NIH below 70
|
really hard to do, talk about giving everyone lipitor
|
|
|
|
why hyperlipidemia
|
smoking (risk for atherosclerosis)
high fat diet diabetic obese hypertensive, high stress in life homocysteine is separate risk factor (if not due to genetic deficiency, then due to low folate and/or vitamin b12 levels (or even low vitamin B6 levels) biggest challenge is patient compliance secondary causes include pregnancy (hypercholesteremia) diabetes (hyperglyceridemia) |
|
|
|
genetic causes of hyperlipidemia
|
LDL receptor def., LDL in bloodstream, cholesterol in bloodstream
abnormal ApoE, more chylomicrons, CM remnants increased synthesis VLDL with decreased breakdown, then VLDL, TGs, lipids increas free no LPL activity or are ApoCII deficient cant break down CMs and VLDL, more TGs in blood |
|
|
|
Tx approaches
|
statin is number one
inhibits HMG-CoA reductase six different kinds of statins side effects include having to monitor liver health/get tests bile acid sequestrants -bile acid is made from cholesterol and goes into intestine to coat lipid droplets in diet helpful with incorporating lipids into cells bile acids are usually recycled, but a bile acid sequestrant blocks this instead they are eliminated into feces body now has to make more bile, thus it pulls cholesterol from bloodstream in order to do so side effects include bloating, constipation, triglycerides increase side effects fibric acid/fibrates ligands of ppar-alpha (peroxisome proliferator activated receptor) increases LPL expression, lowers VLDL -especailly a good thing if have high triglycerides no so well if have high cholesterol side effects include myopathy if combined with statins niacin/nicotinic acid mechanism not known one of bodys vitamins 100x dose of RDA side effects (flushing etc) ezetimbe second part of vytorin keeps lipids fom being absorbed into diet omega 3s PUFA, inhibit TG synthesis raises HDL |
|
|
|
vitamins
|
cannot be synthesized by organism in amount sufficient for physiological needs
absence can cause specific deficiency syndromes |
|
|
|
early 20h century funk purified thiamine, B1 from rice polishing
|
cured beriberi
|
|
|
|
ancient egyptian writings as old as 1500 BC record that liver consumption cured night blindness
|
1747 british physicailn lind discovered that acidic citrus fruits could miraculously cure scurvy
1855 japanese sailors ate polished rice and suffered from beriberi, cured by supplementing with diet of meat, milk, vegetables |
|
|
|
13 vitamins essential for humans
vitamins play many roles in metabolism |
typically used as cofactors for enzymes
-cofactors do post translational modifications to extend range of chemistry that can happen in cell -coenzymes--are attached to enzyme as prosthetic groups, or considered cosubstrates (like NAD) vitamins are organic |
|
|
|
essential nutrients
|
conditionally essential nutrients (choline, lipoic acid, ubiquinone, taurine)
non-essential (that also confer health benefits) -flourine to stabilize teeth -flavonoids for antioxidants, phytoestrogens, carotenoids for antioxidants |
|
|
|
most rapidly dividing cells are ones effected by vitamin deficiency
|
anemia (macrocytic, microcytic)
skin problems CNS problems (BC energy is so critical) |
|
|
|
fat soluble vitamins
|
A D E K
|
|
|
|
NIacin
|
coenzyme is Nicotinamide Adenine Dinucleotide or Nicotinamine Adenine Dinucleotide Phosphate
Redox reactions involving two-electron transfers |
|
|
|
Riboflavin
|
B2
Flavin Mononucleotide Flavin Adenine DiNucleotide one or two electron transfers Redox |
|
|
|
Pantothenate
|
B3
Coenzyme A Transfer of acyl groups |
|
|
|
Thiamine
|
Thiamine Pyrophosphate TPP
transfer of two-carbon groups containing a carbonyl B1 |
|
|
|
pyroxidine
|
Pyroxidal Phosphate (PLP)
B6 Transfer of groups to and from AAs |
|
|
|
Biotin
|
Biotin
ATP dependent carboxylation of substrates or carboxyl group transfer between substrates |
|
|
|
Folate
|
tetrahydrofolate
transfer of one carbon substituents especially formyl and hydroxymethyl groups, provides methyl group for thymine in DNA |
|
|
|
Cobalamin
|
B12
Adenosylcobalamin intramolecular rearrangements Methylcobalamin transfer of methyl groups |
|
|
|
vitmin A
|
retinal, vision
|
|
|
|
vitamin K
|
vitamin K
carboxylation of glutamate residues |
|
|
|
inositol
|
for phospholipids
a sugar alcohol with 6 OH groups important for cell signaling, part of membrane glycoproteins |
|
|
|
Choline
|
in phospholipids, methyl donor, has been demonstrated that healthy men on choline free diet develop liver damage
|
|
|
|
lipoic acid
|
required for pyruvate decarboxylase, animals can make this
|
|
|
|
PQQ
|
pyrroloquinoline quinine (methoxatin)
a redox cofactor for quinoproteins (flavoproteins, dehydrogenases |
|
|
|
Vitamin D
|
synthesized when in sunlight
|
|