Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
295 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
What is the etiology of hemophilia A?
|
x-linked recessive disorder → factor VIII deficiency/inactivity; only symptomatic in males
|
|
|
|
What are examples of hemoglobinopathies?
|
sickle cell anemia
hemoglobin S-C disese sickle cell trait hemoglobin C, D, E, H, I or combination |
|
|
|
What is the etiology of hemophilia B?
|
x-linked recessive disorder → factor IX deficiency/inactivity; only symptomatic in males
|
|
|
|
What is the most common cause of anemia?
|
iron deficiency
|
Current p439
|
|
|
What is hemophilia A?
|
coagulation disorder characterized by factor VIII deficiency/inactivity
|
|
|
|
What are 2 broad categories of macrocytic anemia?
|
megaloblastic anemia
non-megaloblastic anemia |
|
|
|
What is hemophilia B?
|
coagulation disorder characterized by factor IX deficiency/inactivity
|
|
|
|
What are 2 types of megaloblastic anemia?
|
B12 deficiency
folate deficiency |
|
|
|
What is the diagnostic work-up of hemophilia A and B?
|
PT → normal
PTT → prolonged (corrects upon mixing with normal plasma) bleeding time → normal factor VIII → deficiency if hemophilia A factor IX → deficiency if hemophilia B |
|
|
|
What is the definition of megaloblastic anemias?
|
anemias characterized by large RBCs
|
|
|
|
What is the clinical presentation of hemophilia A and B?
|
male
bruising soft-tissue bleeding hemarthrosis spontaneous bleeding involving mucous membranes, skin, joints, muscles, and viscera (if severe) trauma/surgery-associated bleeding (if mild) |
|
|
|
Mucosal changes like smooth tongue indicate what type of anemia?
|
megaloblastic
|
Current p439
|
|
|
What are the complications of hemophilia A and B?
|
recurrent hemarthorses → arthropathy and arthritis
large intramuscular hematomas → compartment syndrome intercranial hemorrhage death from severe bleeding (rare) |
|
|
|
What is the most common cause of non-megaloblastic anemia?
|
alcoholism
|
|
|
|
What is the management of hemophilia A and B?
|
1. refer to hematologist
2. if mild → intranasal or IV DDAVP 2. if severe + child → factor transfusions 2-3 x week 3. if severe + adult → factor transfusions prior to high-risk activities or during bleeding episodes 4. antifibrinolytics for mucosal bleeding 5. celecoxib for arthritis |
Current p491
|
|
|
What is hemodialysis?
|
removal of fluid and waste (creatinine, urea) from blood in patients with chronic kidney disease or renal failure
|
|
|
|
Which is more common, hemophilia A or B?
|
hemophilia A
|
|
|
|
What is the ddx for vitamin B12 deficiency?
|
folate deficiency → indicated by normal B12 and ↓ folate
|
Current p445-446
|
|
|
What is the function of von Willebrand factor?
|
1. forms complex with factor VIII to activate factor X
2. mediates platelet adhesion to extracellular matrix during clot formation |
|
|
|
What are the complications of vitamin B12 deficiency?
|
neurologic symptoms become permanent if not treated within 6 months of onset
|
Current
|
|
|
What is the diagnostic work-up of von Willebrand disease?
|
PT → normal
PTT → prolonged bleeding time → prolonged von willebrand factor → deficiency (though type 2N resembles hemophilia A) |
|
|
|
What is the management for vitamin B12 deficiency?
|
prescribe vitamin B12 → 100mcg IM daily first week, weekly first month, monthly for life; 1000mcg PO 1x per day for life
|
Current
|
|
|
What is the etiology of von Willebrand disease?
|
autosomal dominant disorder → von Willebrand factor deficiency
|
|
|
|
What is the patient education for vitamin B12 deficiency?
|
vitamin B12 present in dairy, eggs, fish, poulty, and meat; vitamin B12 supplementation is required for life
|
Current
|
|
|
What is von Willebrand disease?
|
coagulation disorder characterized by von Willebrand factor deficiency
|
|
|
|
What is the etiology of folate deficiency anemia?
|
1. inadequate dietary intake (poor nutrition, overcooked food, alcoholics, anorexics)
2. increased dietary requirement (pregnancy, exfoliative skin disease, chronic hemolytic anemia) 3. inadequate absorption (tropical sprue, drug interaction) 4. loss (hemodialysis) 5. folate antagonist (methotrexate) |
Current
|
|
|
What is the clinical presentation of von Willebrand disease?
|
easy bruising and epistaxis from early childhood
menorrhagia prolonged bleeding following trauma/surgery |
|
|
|
What is the patient education for folate deficiency anemia?
|
folate present in most fruits and vegetables (especially citrus fruits and green leafy vegetables)
|
Current
|
|
|
What is the management of von Willebrand disease?
|
1. if mild → desmopressin acetate
2. if severe → vWF-containing factor VIII transfusions 3. antifibrinolytics for mucosal bleeding 4. estrogen-containing contraceptives for menorrhagia 5. avoid aspirin and NSAIDs |
|
|
|
What is aplastic anemia?
|
failure of bone marrow to produce RBCs, WBCs, or platelets
|
|
|
|
What is the etiology of aplastic anemia?
|
either direct stem cell injury or autoimmune disease mediated by T-cells or IgG Ab against stem cells; usually autoimmune
|
|
|
|
If the results of an automated DIFF are abnormal, how are they verified?
|
via manual DIFF
|
Interpreting Laboratory Data p340
|
|
|
What is the ddx for aplastic anemia?
|
idiopathic → probably autoimmune
congenital → rare pregnancy posthepatitis paroxysmal nocturnal hemoglobinuria SLE toxins → benzene, toluene, insecticides medications → phenytoin, carbamazepine, sulfonamides, chloramphenicol, pehnylbutazone, gold salts, quinacrine, tolbutamide chemotherapy radiation |
Current p454
|
|
|
What is the ddx for aplastic anemia?
|
idiopathic → probably autoimmune
congenital → rare pregnancy posthepatitis paroxysmal nocturnal hemoglobinuria SLE toxins → benzene, toluene, insecticides medications → phenytoin, carbamazepine, sulfonamides, chloramphenicol, pehnylbutazone, gold salts, quinacrine, tolbutamide chemotherapy radiation |
Current p454
|
|
|
What is the clinical presentation of aplastic anemia?
|
anemia → fatigue, weakness, pallor
neutropenia → bacterial infection thrombocytopenia → skin and mucosal bleeding, petechiae, purpura *hepatosplenomegaly, lymphadenopathy, and bone tenderness should NOT be present |
Current p454
|
|
|
What is the diagnostic work-up of aplastic anemia?
|
CBC → pancytopenia
RETIC → low blood smear → normal morphology bone marrow biopsy → hypocellular, normal morphology |
Current p454
|
|
|
What are 3 broad etiologies of anemia?
|
1. blood loss
2. increased destruction of RBCs 3. impaired production of RBCs |
hematology lecture
|
|
|
What is the management of aplastic anemia?
|
1. if mild, supportive care as necessary → RBC and platelet transfusions, antibiotics
2. if severe + <50y/o → bone marrow transplant from HLA-matched sibling donor 3. if severe + >50y/o or no HLA-matched sibling donor → ATG + cyclosporine |
Current p455
|
|
|
Increased RETIC is indicative of?
|
blood loss
destruction of RBCs - hemolytic anemia EPO treatment |
Antrim
|
|
|
What is the prognosis for aplastic anemia?
|
1. if severe and untreated → 3 months to live
2. if bone marrow transplant → 80% success rate 3. if ATG → 60% success rate; improvement within 4-12 weeks; usually only partial response |
Current p455
|
|
|
Decreased RETIC is indicative of?
|
impaired production of RBCs
-nutritional deficiency - iron, B12, folate -marrow empty - aplastic anemia, pure red cell aplasia -marrow asleep - EPO deficiency - anemia of chronic disease, renal failure -marrow broken - myelodysplasia -marrow full of something else - infection, cancer |
hematology lecture
|
|
|
What bone tumor presents at the diaphysis?
|
Ewing's sarcoma
|
Orthopedics p448
|
|
|
List the ddx for macrocytic anemia?
|
MEGALOBLASTIC:
B12 deficiency folate deficiency NON-MEGALOBLASTIC: liver disease kidney disease reticulocytosis myelodysplasia anti-retrovirals |
|
|
|
Sickle cell anemia imparts resistance to what disease?
|
malaria
|
|
|
|
List the indications for prescribing EPO?
|
anemia due to chronic kidney disease
myelodysplasia |
|
|
|
SICKLE CELL ANEMIA:
|
ETIOLOGY:
homozygous genetic disorder characterized by hemoglobin S (instead of hemoglobin A) common in African, Mediterranean, Middle Eastern, Indian, or Caribbean CLINICAL PRESENTATION: fatigue, pallor, jaundice dactylitis, recurrent abdominal or musculoskeletal pain gallstones, splenomegaly in early childhood with later disappearance DIAGNOSTIC WORKUP: NBS CBC → normocytic or macrocytic anemia peripheral blood smear → sickle cells, target cells (hemolytic anemia) ↑ RETIC hemoglobin electrophoresis MANAGEMENT: education program prophylactic penicillin started at 2m/o and continued until at least 5y/o immunizations if illness with fever >38.5°C → BC, IV broad-spectrum antibiotics, observation if vaso-occlusive episode → maintain adequate analgesia, hydration, and oxygen saturation; correct acidosis; treat infections if acute severe exacerbation of anemia (splenic sequestration, aplastic crisis), acute severe vaso-occlusive episode (acute severe chest syndrome, stroke, organ failure), high-risk procedures, surgery → packed RBCs *acute chest syndrome = fever, pleuritic chest pain, pulmonary infiltrates with hypoxemia caused by pulmonary infection, infarction, or fat embolism from ischemic bone marrow hydroxyurea PROGNOSIS: early identification and prophylactic penicillin allow children to live into adulthood but end-organ damage d/t vaso-occlusion invariably occurs |
|
|
|
What is the etiology of B12 deficiency?
|
malabsorption
-lack of stomach acid -lack of intrinsic factor -lack of pancreatic enzymes -lack of bowel -something else eating B12 dietary deficiency rare |
anemia lecture
|
|
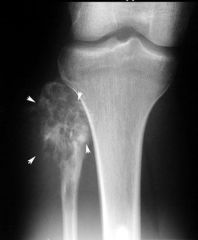
|
bone destruction + sunburst pattern → osteosarcoma (of proximal fibula)
|
|
|
|
What is immune thrombocytopenic purpura (ITP)?
|
autoimmune disorder characterized by low platelet count
|
Current p482
|
|
|
What methylmalonic acid and homocysteine results are seen in B12 deficiency?
|
both are increased
|
anemia lecture
|
|
|
What is the etiology of ITP?
|
autoimmune disease → antibodies bind platelets → accelerated platelet clearance; usually primary and idiopathic
often follows infection with viruses, such as rubella, varicella, measles, parvovirus, influenza, or EBV usually occurs in children 2-5y/o |
Current p482
|
|
|
What methylmelonic acid and homocysteine results are seen in folate deficiency?
|
homocysteine increased
(no link between methylmelonic acid and folate deficiency) |
anemia lecture
|
|

|
Ewing's sarcoma (of femur)
|
|
|

|
Ewing's sarcoma (of femur)
|
|
|
|
What is hemolytic anemia?
|
anemia caused by destruction of RBCs
|
|
|
|
What is intrinsic hemolytic anemia?
|
anemia due to defect inside RBCs that results in hemolysis; defects include abnormal hemoglobin, abnormal RBC cell wall, abnormal RBC metabolism
|
|
|
|
What is extrinsic hemolytic anemia?
|
anemia due to factors outside RBCs that result in hemolysis; factors include autoimmune diseases, drugs
|
|
|
|
What are examples of intrinsic and extrinsic hemolytic anemia?
|
Intrinsic:
sickle cell anemia hemoglobin S-C disease sickle cell trait hereditary spherocytosis G6DP deficiency Extrinsic: autoimmune hemolytic anemia cold agglutinin disease |
|
|
|
What is anemia of chronic disease?
|
microcytic hypochromic or normocytic normochromic anemia associated with chronic disease
|
Pathology p78
|
|
|
What is the etiology of anemia of chronic disease?
|
associated with chronic infection, chronic inflammation, liver disease, kidney disease, cancer, inadequate dietary intake due to illness, hemodialysis
|
Pathology p78
|
|
|
What is the diagnostic work-up of anemia of chronic disease?
|
CBC → low RBC, hct, hgb; normal or low MCV
FERR → normal or high IRON → possibly low TIBC → low |
Pathology p78
|
|
|
Define -cytosis.
|
increased
|
Stedmans
|
|
|
Define -cytopenia.
|
decreased
|
Stedmans
|
|
|
Define leukocytosis.
|
abnormally high WBC count
|
Stedmans
|
|
|
Define leukocytopenia (AKA leukopenia).
|
abnormally low WBC count
|
Stedmans
|
|
|
Define erythrocytosis (AKA polycythemia).
|
abnormally high RBC count
|
Stedmans
|
|
|
Define erythrocytopenia (AKA erythropenia, hypocythemia, anemia).
|
abnormally low RBC count
|
Stedmans
|
|
|
Define thrombocytosis.
|
abnormally high platelet count
|
Stedmans
|
|
|
Define thrombocytopenia (AKA thrombopenia).
|
abnormally low platelet count
|
Stedmans
|
|
|
Define pancytopenia.
|
abnormally low WBCs, RBCs, and platelets
|
Stedmans
|
|
|
Define anisocytosis.
|
abnormal variation in size of cells that are normally uniform in size
*usually refers to RBCs where abnormal variation in size of RBCs is indicated by abnormally high RDW in CBC |
|
|
|
Define poikilocytosis.
|
abnormally shaped RBCs
|
|
|
|
Define reticulocytosis.
|
abnormally high retic count
|
|
|
|
Define hemoglobinopathy.
|
disorder associated with abnormal hemoglobin
|
Stedmans
|
|
|
Define hemolysis.
|
alteration or destruction of RBCs causing hemoglobin to be released into medium in which cells are suspended; may be caused by abnormal RBC structure, antibodies, temperature, trauma, tonicity, chemicals, toxins
|
Stedmans
|
|
|
List symptoms of anemia.
|
fatigue
weakness dizziness syncope hypotension tachycardia skin pallor jaundice → hemolytic anemia cold skin conjunctival pallor hair loss brittle nails conjunctival icterus → hemolytic anemia cheilits glossitis dyspnea palpitations chest pain splenomegaly → hemolytic anemia lymphadenopathy hepatosplenomegaly bone tenderness |
|
|
|
What are 3 broad categories of anemia?
|
microcytic
macrocytic normocytic |
|
|
|
List ddx of microcytic anemia.
|
iron deficiency
lead poisoning anemia of chronic disease thalassemia sideroblastic anemia hemoglobinopathies |
Current p439
hematology lecture |
|
|
What is the diagnostic work-up for iron deficiency anemia?
|
CBC → low RBC, hct, hgb, MCV
FERR → low IRON → low TIBC → high (empty binding sites on transferrin due to lack of iron to transport) |
|
|
|
What is the most important lab test to order for suspected iron deficiency anemia?
|
FERR
|
|
|
|
What FERR level rules out iron deficiency?
|
>100 ng/dL
|
anemia lecture
|
|
|
What is the management for iron deficiency anemia?
|
1. prescribe ferrous sulfate 325mg PO 3x per day for 3-6 months
2. monitor CBC, FERR, IRON, TIBC |
|
|
|
After treating iron deficiency with ferrous sulfate, how long before RETIC increases, Hct increases, and RBC count normalizes?
|
RETIC increases in 7-10 days; Hct increases in 2-3 weeks; RBC count normalizes in 2 months
|
hematology lecture
|
|
|
List possible causes for non-response to ferrous sulfate for iron deficiency anemia and how to rectify them.
|
non-compliance → prescribe lower dose or take iron with food
blood loss → determine cause defective absorption → prescribe concomitant ascorbic acid 250mg PO daily concurrent B12 or folate deficiency wrong diagnosis |
anemia lecture
|
|
|
What is hematopoiesis?
|
synthesis of formed elements (WBCs, RBCs, and platelets)
|
|
|
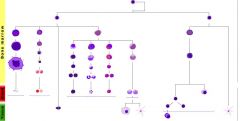
Describe the development of cell lines in hematopoiesis.
|
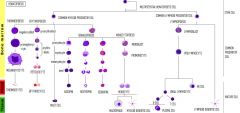
|
|
|
|
What is the lifespan of mature RBCs?
|
120 days
|
Interpreting Laboratory Data p342
|
|
|
How are RBCs removed from circulation?
|
by macrophages from spleen
|
Interpreting Laboratory Data p342
|
|
|
What are reticulocytes?
|
immature nucleated RBCs
|
|
|
|
Where are erythropoietin and thrombopoietin synthesized?
|
liver and kidneys
|
|
|
|
What is haptoglobin?
|
plasma protein that binds and clears hemoglobin released into plasma
|
Current p447
|
|
|
Where does hematopoiesis occur in adults?
|
red bone marrow of skull, ribs, vertebrae, pelvis, humerus, and femur
|
Pathology p72
|
|
|
Where does hematopoiesis occur in children?
|
bone marrow of long bones (femur, tibia)
|
|
|
|
What is erythropoietin (EPO)?
|
hormone that stimulates myeloid progenitor cells to differentiate into erythrocytes
|
|
|
|
What is thrombopoietin (TPO)?
|
hormone that stimulates myeloid progenitor cells to differentiate into platelets
|
|
|
|
What is extramedullary hematopoiesis?
|
if normal hematopoiesis compromised, blood cells can be produced by liver, spleen, and lymph nodes
|
Pathology p72
|
|
|
What is koilonychia?
|
spoon-shaped nails caused by lack of adequate tissue oxygenation
|
Pathology p72
|
|
|
What are spherocytes and their associated causes?
|
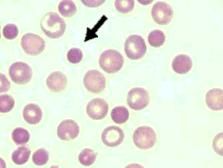
RBCs that are sphere-shaped, hyperchromic, no central pallor; associated with hereditary spherocytosis, immune hemolytic anemia
|
Pathology p72
|
|
|
What are schistocytes (helmet cells) and their associated causes?
|
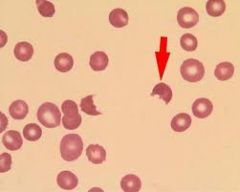
RBC fragments; associated with microangiopathic hemolytic anemias
|
Pathology p72
|
|
|
What are target cells and their associated causes?
|
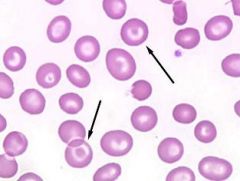
RBCs with rim of pallor around normochromic core; associated with thalaseemia, liver disease, hemoglobin C disease
|
Pathology p72
|
|
|
What hematocrit level indicates anemia?
|
<41% for men
<37% for women |
Current p439
|
|
|
What hemoglobin level indicates anemia?
|
<13.5 g/dL for men
<12.0 g/dL for women |
Current p439
|
|
|
What are the components of a CBC?
|
WBC count
RBC count hct hgb MCV MCH MCHC RDW platelet count + MPV |
Interpreting Laboratory Data p340
|
|
|
What is the RBC count?
|
number of RBCs in a given volume of blood
|
Interpreting Laboratory Data p340
|
|
|
What is the normal adult range for RBCs?
|
males 4.5-5.9 x10^6 cells/microliter
females 4.1-5.1 x 10^6 cells/microliter |
Interpreting Laboratory Data p340
|
|
|
What are 2 reasons women have lower RBC counts than men?
|
1. menstrual blood loss
2. lower concentrations of androgen (erythropoietic stimulant) than men |
Interpreting Laboratory Data p340
|
|
|
What CBC results indicate microcytic anemia?
|
↓ RBC count + ↓MCV
|
|
|
|
What CBC results indicate macrocytic anemia?
|
↓ RBC count + ↑ MCV
|
|
|
|
What CBC results indicate normocytic anemia?
|
↓ RBC count + normal MCV
|
|
|
|
MCV values are higher in newborns/infants than adults, true or false?
|
true
|
hematology lecture
|
|
|
What is hypochromic?
|
abnormally low MCH/MCHC where RBCs appear lighter than normal
|
|
|
|
What is hematocrit?
|
% of RBCs in given volume of blood
|
|
|
|
What are the normals for MCV?
|
80-100 mm³
|
|
|
|
MCV <80 mm³ indictes?
|
microcytic anemia
|
|
|
|
MCV >100 mm³ indicates?
|
macrocytic anemia
|
|
|
|
What is hyperchromic?
|
abnormally high MCH/MCHC where RBCs appear darker than normal
|
|
|
|
How is iron stored?
|
as ferritin or hemosiderin
|
|
|
|
What is iron deficiency anemia?
|
microcytic hypochromic anemia characterized by low iron
|
|
|
|
Where does iron absorption occur?
|
stomach, duodenum, upper jejunum
|
|
|
|
What is transferrin?
|
plasma protein that transports iron
|
|
|
|
What does TIBC stand for?
|
total iron binding capacity
|
|
|
|
What is the clinical presentation of iron deficiency anemia?
|
fatigue, pallor, tachycardia, tachypnea on exertion, palpitations; if severe, hair loss, cheilitis, glossitis, spooned nails
|
|
|
|
What is the etiology of iron deficiency anemia?
|
1. nutritional deficiency → inadequate intake (poor, alcoholics), increased requirements (pregnancy)
2. malabsorption 3. iron sequestration 4. hemolysis 5. blood loss |
|
|
|
What is the physical exam for supsected anemia?
|
1. heart rate for tachycardia
2. respirations for tachypnea 3. skin exam for hair loss, pallor, brittle nails 4. eye exam for conjunctival pallor 5. oral exam for cheilitis, glossitis 6. cardiac exam for palpitations 7. rectal exam for blood loss |
|
|
|
What is the management of anemia of chronic disease?
|
1. usually treatment unecessary
2. if transfusion-dependent or quality of life improved by response → prescribe EPO *EPO expensive |
|
|
|
What is vitamin B12 deficiency anemia?
|
macrocytic megaloblastic anemia characterized by low vitamin B12
|
|
|
|
What is the etiology of vitamin B12 deficiency?
|
1. inadequate dietary intake
2. malabsorption → blind loop syndrome, tapeworm, Crohn's disease, surgery rare; common in vegans |
|
|
|
What is the clinical presentation of vitamin B12 deficiency anemia?
|
onset >3 years after absorption ceases due to large liver stores; fatigue, tachycardia, tachypnea on exertion, pallor, glossitis, anorexia, diarrhea, paresthesias, impaired balance
|
|
|
|
What does AML stand for?
|
acute myeloid leukemia
|
|
|
|
Anemia + schistocytes indicates?
|
microangiopathic hemolytic anemia
|
|
|
|
What are examples of microangiopathic hemolytic anemia?
|
hemolytic uremic syndrome
DIC thrombotic thrombocytopenic purpura aortic stenosis malfunctioning cardiac valve prosthesis |
|
|
|
What is the pathophysiology of schistocyte formation?
|
damage to blood vessel endothelium → fibrin deposition and platelet aggregation → RBCs fragmented as travel through obstructed blood vessel → intravascular hemolysis
|
|
|
|
What is microangiopathic hemolytic anemia?
|
group of intravascular hemolytic anemias characterized by schistocytes
|
|
|
|
What does ATG stand for and what is it used for?
|
antithymocyte globulin; immunosuppresive therapy
|
Current p447
|
|
|
What is the ddx for pancytopenia?
|
marrow disorders:
megaloblastic anemia aplastic anemia myelodysplasia acute leukemia myelofibrosis myeloma lymphoma hairy cell leukemia → splenomegaly, bone marrow biopsy reveals abnormal lymphoid cells carcinoma non-bone marrow disorders: hypersplenism TB AIDS leishmaniasis brucellosis SLE |
Current p455
|
|
|
What is polycythemia vera?
|
acquired myeloproliferative disease characterized by increased RBC count
|
|
|
|
What is the diagnostic work-up of polycythemia vera?
|
WBC → normal or increased
RBC → increased platelet count → normal or increased blood smear → no abnormal cells seen JAK2 → mutation present |
|
|
|
What is the management for polycythemia vera?
|
1. phlebotomy → 500mL (1 unit) removed weekly until hct <45%
2. phlebotomy as necessary to maintain hct <45% |
|
|
|
How long are autologous packed RBCs stored?
|
35 days
|
Current p477
|
|
|
What is pure red cell aplasia?
|
autoimmune disease characterized by failure of bone marrow to produce RBCs
|
|
|
|
What is the etiology of pure red cell aplasia?
|
autoimmune disease mediated by T-cells or rarely IgG Ab against erythocyte precursor cells; usually idiopathic
|
Current p447
|
|
|
What is the ddx for pure red cell aplasia?
|
usually idiopathic
SLE CLL lymphoma thymoma phenytoin chloramphenicol if transient → viral infection → parvovirus |
Current p447
|
|
|
What is the clinical presentation of pure red cell aplasia?
|
anemia → fatigue, weakness, pallor
|
Current p447
|
|
|
What is the diagnostic work-up of pure red cell aplasia?
|
CBC → severe normochromic anemia
RETIC → low or absent blood smear → RBC morphology normal bone marrow biopsy → normocellular, erythrocyte precursors low or absent |
Current p447
|
|
|
What is the management of pure red cell aplasia?
|
1. refer to hematologist
2. immunosuppressive therapy → ATG + cyclosporine 3. resection if thymoma 4. stop medications if drug-related |
Cureent p447
|
|
|
How do you distinguish pure red cell aplasia, aplastic anemia, and myelodysplasia?
|
pure red cell aplasia → anemia, normal morphology, normocelluar bone marrow
aplastic anemia → pancytopenia, normal morphology, hypocellular bone marrow myelodysplasia → abnormal morphology |
Current p447
|
|
|
What is polycythemia vera?
|
acquired bone marrow (myeloproliferative) disorder characterized by abnormally high hct
|
Current p457
|
|
|
What is the etiology of polycythemia vera?
|
acquired overproduction of RBCs (independent of EPO) due to JAK2 mutation; rare if <40y/o
|
Current p457
|
|
|
What is the ddx for polycythemia?
|
spurious polycythemia → low plasma volume
primary polycythemia: polycythemia vera secondary polycythemia: hypoxia → cardiac, pulmonary, high altitude carboxyhemoglobin → smoking kidney lesions EPO-secreting tumor → rare abnormal hgb → rare |
Current p458
|
|
|
What is the clinical presentation of polycythemia vera?
|
increased blood volume and viscosity → fatigue, HA, dizziness, blurred vision, tinnitus
increased basophils → generalized pruritus (especially following warm shower) engorgement of mucosal blood vessels → epistaxis engorged retinal veins splenomegaly |
Current p457
|
|
|
What is the diagnostic work-up of polycythemia vera?
|
CBC → high hct, WBC count, and platelet count (but hct count especially)
blood smear → normal morphology B12 → high EPO → low JAK2 → mutation present blood gas → normal O2 |
Current p457
|
|
|
What is management of polycythemia vera?
|
1. phlebotomy → 500mL (1 unit) removed weekly until hct <45% then as necessary for maintenance of hct <45%
2. aspirin for reduced risk of thrombosis → 75-81mg daily 3. antihistamine for pruritus→ diphenhydramine 4. low iron diet 5. if phlebotomy problematic → hydroxyurea 6. if excessive phlebotomy, thrombocytosis, or pruritus → myelosuppressive therapy |
Current p458
|
|
|
What are the complications of polycythemia vera?
|
1. thrombosis → major cause of morbidity and death
2. bleeding → may lower hct creating diagnostic confusion; may cause iron deficiency anemia 3. peptic ulcer disease |
Current p457
|
|
|
Define plethora.
|
an excess of blood in circulatory system
|
|
|
|
Define hypersplenism.
|
disorder characterized by premature and rapid destruction of RBCs by spleen
|
|
|
|
What is the ddx for JAK2 mutation?
|
polycythemia vera
myelofibrosis essential thrombocytosis |
Current p458
|
|
|
What is essential thrombocytosis?
|
idiopathic myeloproliferative disorder characterized by abnormally high platelet count
|
Current p459
|
|
|
What is the etiology of essential thrombocytosis?
|
proliferation of mekakaryocytes in bone marrow causing high platelet count; idiopathic but JAK2 mutation usually present
|
Current p459
|
|
|
What is the clinical presentation of essential thrombocytosis?
|
high platelet count
thrombosis erythromelalgia → erythema + painful burning of hands splenomegaly → 25% of patients mucosal bleeding → if concomitant platelet defect |
Current p459
|
|
|
What is the diagnostic work-up of essential thrombocytosis?
|
CBC → high platelet count, mildly elevated WBC count
blood smear → large platelets (but not giant degranulated forms) bone marrow biopsy → increased megakaryocytes philadelphia chromosome → absent (present in CML) |
Current p459
|
|
|
What is the management of essential thrombocytosis?
|
1. refer to hematologist
2. hydroxyurea to reduce platelet count to <500,000/mcL → 0.5-2g/d 3. add anagrelide if anemia due to hydroxyurea → 1-2mg/d 4. aspirin for erythromelalgia and to reduce risk of thrombosis → 81mg/d 5. if severe bleeding → plateletpheresis to rapidly lower platelet count |
Current p459
|
|
|
What are the complications of essential thrombocytosis?
|
thrombosis
late in course of disease → massive splenomegaly, splenic infarction, fibrotic bone marrow myelofibrosis → 10-15% risk after 15 years acute leukemia → 1-5% risk over 20 years |
Current p459
|
|
|
What is the ddx for thrombocytosis?
|
essential thrombocytosis → platelet count often >200,000/mcL
reactive thrombocytosis → platelet count rarely >100,000/mcL chronic infection inflammatory disorders → RA, ulcerative colitis splenectomy → temporary polycythemia vera myelofibrosis CML |
Current p459
|
|
|
How do you distinguish essential thrombosis, myelofibrosis, and CML?
|
essential thrombosis → normal RBC morphology, large platelets (but giant degranulated platelets absent), philadelphia chromosome absent
myelofibrosis → abnormal RBC morphology, giant degranulated platelets CML → philadelphia chromosome present |
Current p459
|
|
|
What is myelofibrosis?
|
myeloproliferative disorder characterized by bone marrow fibrosis
|
Current p460
|
|
|
What is the etiology of myelofibrosis?
|
increased secretion of platelet-derived growth factor (PDGF) and other cytokines; JAK2 mutation may be present
|
Current p460
|
|
|
What is the clinical presentation of myelofibrosis?
|
insidious onset
anemia → fatigue massive splenomegaly |
Current p460
|
|
|
What is the diagnostic work-up of myelofibrosis?
|
1. CBC → anemia, variable WBC and platelet counts
2. blood smear → teardrop poikilocytosis, leukoerythroblasts, giant degranulated platelets 3. bone marrow aspiration usually results in dry tap but if performed → hypocelluar, increased megakarytocytes 4. silver stain or bone marrow biospy → fibrosis |
Current p460
|
|
|
What is the management of myelofibrosis?
|
1. refer to hematologist
2. if mild → no treatment 3. if anemia → blood transfusion or EPO 3. if young → bone marrow transplant 4. thalidomide or lenalidomide |
Current p460
|
|
|
What are the complications of myelofibrosis?
|
thrombocytopenia → bleeding
splenomegaly → early satiety, splenic infarction bone pain (especially upper legs) hematopoiesis in liver → portal HTN, ascites, esophageal varices |
Current p460
|
|
|
What is the ddx for bone marrow fibrosis?
|
myelofibrosis
metastatic carcinoma Hodgkin disease hairy cell leukemia |
Current p460
|
|
|
What is the prognosis for myelofibrosis?
|
5 year survival rate
|
Current p461
|
|
|
What triad usually indicates myelofibrosis?
|
teardrop poikilocytosis
leukoerythroblasts giant degranulated platelets |
Current p461
|
|
|
What does CML stand for?
|
chronic myeloid leukemia
|
|
|
|
What is CML?
|
myeloproliferative disorder characterized by overproduction of myeloid cells with normal bone marrow function in early phase
|
Current p461
|
|
|
What is the etiology of CML?
|
philadelphia chromosome; presents during middle age
|
Current p461
|
|
|
What is the clinical presentation of CML?
|
fatigue, low fever, night sweats
splenomegaly sternal tenderness due to marrow overexpansion |
Current p461
|
|
|
What is the diagnostic work-up of CML?
|
CBC → high WBC count
blood smear → shift to the left, dominated by mature forms bone marrow biopsy → hypercelluar, shift to the left PCR for philadelphia chromosome → bcr/abl gene present |
Current p461
|
|
|
What is the management of CML?
|
1. refer to hematologist
If normal bone marrow function: 2. imatinib mesylate 400mg PO daily 3. if refractory to oral agents → bone marrow transplant If accelerated CML: 4. imatinib 600mg PO daily initially 5. bone marrow transplant 7. if leukostasis → emergent leukophoresis + myelosuppressive therapy |
Current p462
|
|
|
What are the complication sof CML?
|
1. leukostasis
2. progression to acute leukemia |
Current p462
|
|
|
What is the prognosis of CML?
|
80% survival rate at 7 years
|
Current p462
|
|
|
List myeloproliferative disorders.
|
polycythemia vera
essential thrombocytosis myelofibrosis CML |
|
|
|
What is leukostasis.
|
abnormal intravascular leukocyte aggregation and clumping; complication of leukemia
|
|
|
|
What is the clinical presentation of leukostasis?
|
blurred vision, respiratory distress, priapism
|
Current p462
|
|
|
Recurrent bleeding in children at multiple sites may indicate?
|
heritable hemostasis disorder
|
Current p481
|
|
|
What are examples of coagulopathies?
|
hemophilia A
hemophilia B Von Willebrand disease vitamin K deficiency liver disease Dengue Fever leukemia DIC |
|
|
|
What is the function of von Willebrand factor?
|
1. forms complex with factor VIII to activate factor X
2. mediates platelet adhesion to extracellular matrix during clot formation |
|
|
|
What is the clinical presentation of a platelet disorder?
|
bleeding localized to skin and mucous membranes → petechiae, purpura, ecchymosis, epistaxis, gingival bleeding
small superficial nonpalpable ecchymosis bleeding after minor wounds immediate post-surgical bleeding |
hemostasis lecture
|
|
|
What symptoms/signs characterize coagulation disorders?
|
bleeding localized to joints and muscles
no petechiae large palpable ecchymosis bleeding after minor wounds rare delayed post-surgical bleeding |
hemostasis lecture
|
|
|
Does PT measure intrinsic or extrinsic coagulation pathway?
|
extrinsic
|
hemostasis lecture
|
|
|
Does PTT measure intrinsic or extrinsic coagulation pathway?
|
intrinsic
|
hemostasis lecture
|
|
|
Prolonged PTT indicates a possible deficiency in what factors?
|
XII, XI, IX, VIII, von willebrand, X, V, II, I
|
hemostasis lecture
|
|
|
Prolonged PT indicates a possible deficiency in what factors?
|
VII, X, V, II, I
|
hemostasis lecture
|
|
|
What is the most common cause of elevated PTT?
|
von Willebrand disease
|
hemostasis lecture
|
|
|
Define coagulopathy.
|
disorder associated with abnormal coagulation
|
Stedmans
|
|
|
Bleeding time is a measure of?
|
platelet function
|
|
|
|
What is the diagnostic work-up of hemophila B?
|
PT → normal
PTT → prolonged bleeding time → normal factor IX → deficiency |
|
|
|
What is the most common hereditary bleeding disorder?
|
von Willebrand disease
|
|
|
|
What are the steps of the extrinsic pathway?
|
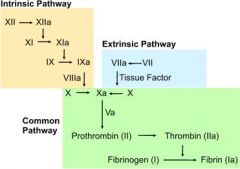
1. tissue injury → release of tissue factor (factor III)
2. tissue factor activates factor VII 3. factor VIIa activates factor X |
|
|
|
What are the steps of the intrinsic pathway?
|
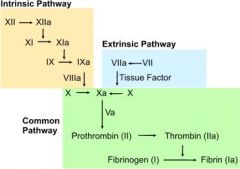
1. exposed collagen following tissue injury activates factor XII
2. factor XIIa activates factor XI 3. factor XIa activates factor IX 4. factor IXa + factor VIIIa activate factor X |
|
|
|
What are the steps of the common coagulation pathway?
|
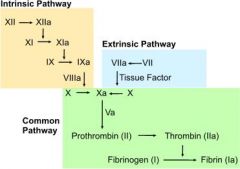
1. Xa + Va catalyzes prothrombin (factor II) → thrombin
2. thrombin catalyzes fibrinogen (factor I) → fibrin 3. fibrin → clot |
|
|
|
What is the function of antithrombin?
|
inactivates thrombin
|
|
|
|
How does heparin act as an anticoagulant?
|
1. heparin activates antithrombin
2. antithrombin inactivates thrombin |
|
|
|
What clotting factors and anticoagulation proteins are vitamin K dependent?
|
clotting factors → VII, IX, X, II
anticoagulation proteins → protein S, protein C |
|
|
|
How does warfarin act as an anticoagulant?
|
1. vitamin K epoxide reductase necessary to convert glu to gla (part of clotting factor)
2. gla necessary for clotting factors to stably bind to blood vessel endothelium 3. stable binding necessary to activate clotting 4. warfarin inhibits vitamin K epoxide reductase → lower concentration of viable clotting factors |
|
|
|
What are clotting factors?
|
plasma proteins involved in the clotting cascade
|
|
|
|
What are the functions of thrombin?
|
1. catalyzes fibrinogen → fibrin
2. activates factor VIII and factor V 3. stimulates platelet aggregation 4. activates protein C |
|
|
|
What is the function of protein C?
|
1. activated by thrombin + thrombomodulin
2. inactivates factor VIII and factor V 3. increases fibrinolysis |
|
|
|
What is the function of protein S?
|
acts as cofactor to protein C; increases rate at which protein C inactivates factor Va
|
|
|
|
What is the evaluation of a bleeding disorder?
|
1. onset
childhood → heritable disorder adult → mild heritable disorder or acquired 2. location mucocutenous → platelet disorder joints and muscles → clotting factor disorder 3. clinical context → pregnancy, underlying medical conditions, sepsis, medications, postsurgery 4. personal history → prior spotaneous bleeding, excessive bleeding with trauma, dental, menses, surgery 5. family history |
Current p481
|
|
|
What is the ddx for thrombocytopenia (<150,000/mcL)?
|
Decreased synthesis of platelets:
nutritional deficiency → iron , B12, folate, alcohol congenital bone marrow failure acquired bone marrow failure → aplastic anemia, myelodysplasia infiltrated bone marrow → infection, leukemia, metastatic carcinoma chemotherapy radiation Increased destruction of platelets: immune thrombocytopenic purpura thrombotic thromboctypenic purpura hemolytic uremic syndrome DIC Increased sequestration of platelets: hypersplenism |
|
|
|
What is the etiology of ITP?
|
autoimmune disease → antibodies bind platelets → accelerated platelet clearance; usually primary and idiopathic
|
Current p482
|
|
|
What is the clinical presentation of ITP?
|
mucocutaneous bleeding
petechiae, ecchymosis |
Current p482
|
|
|
What is the diagnostic work-up of ITP?
|
CBC → anemia if bleeding, thrombocytopenia
PT → normal PTT → normal bleeding time → prolonged HepC HIV |
Current p482
|
|
|
What is the management of ITP?
|
1. refer to specialist
2. d/c offending medications 3. if >60y/o, unexplained cytopenia, or non-response to treatment → bone marrow biopsy → normocellular, normal megakaryocyte morphology 4. if mild → observe 5. if platelet count <20,000/mcL or significant bleeding → corticosteroids + IV immunoglobulin 6. if hemorrhage → platelet transfusion |
Current p483
|
|
|
What are the complicatons of ITP?
|
intercranial hemorrhage
|
Pathology p83
|
|
|
What is the prognosis of ITP?
|
relapse common
|
Current p483
|
|
|
What is the ddx for ITP?
|
primary:
idiopathic secondary: hepatitis C HIV lupus lymphoma medications |
Current p482
|
|
|
What does ITP stand for?
|
immune thrombocytopenic purpura
|
|
|
|
What does TTP stand for?
|
thrombotic thrombocytopenic purpura
|
|
|
|
What is thrombotic thrombocytopenic purpura (TTP)?
|
disorder characterized by thrombi in vasculature, thrombocytosis, microangiopathic hemolytic anemia, and neurological symptoms
|
Current p485
|
|
|
What is the etiology of TTP?
|
inherited or acquired enzyme deficiency in von Willebrand cleaving protease → formation of thrombi → adherence of platelets to thrombi → causing thrombocytopenia + microangiopathic hemolytic anemia
|
Current p485
|
|
|
What is the clinical presentation of TTP?
|
mucocutaneous bleeding → purpura
fever neurological symptoms → HA, drowsiness, delirium, seizures, paresis, coma (due to thrombi in cerebral vasculature) renal failure |
Current p485
|
|
|
What is the diagnostic work-up fo TTP?
|
CBC → anemia, thrombocytopenia
RETIC → high blood smear → schistocytes indirect BILI → high LD → high CREAT → high PT → normal PTT → normal bleeding time → prolonged vWFCP → deficiency |
Current p485
|
|
|
What is the managment of TTP?
|
1. hospitilize and refer to specialist
2. immediant plasma exchange → daily until normal platelet count and LD for 2 days then taper 3. if refractory → plasma exchange twice daily 4. if immediate plasma exchange unavailable → FFP transfusion 5. if anemia → RBC transfusion 6. if renal failure → hemodialysis 7. if life-threatening bleeding → platelet transfusion (otherwise contraindicated) |
Current p485
|
|
|
What are the complications of TTP?
|
1. thrombi in brain → neurological problems, coma
2. if untreated → organ failure → death |
Current p485
|
|
|
What is the prognosis of TTP?
|
95% mortality rate if untreated
|
Current p485
|
|
|
What is the ddx for TTP?
|
genetic → enzyme deficiency of protease that degrades vWF
acquired → antibody prevents protease from degrading vWF HIV cancer medications bone marrow transplant |
Current p485
|
|
|
What is the pentad of thrombotic microangiopathy (TTP or HUS)?
|
1. thrombocytopenia → both
2. microangiopathic hemolytic anemia → both 3. fever → usually TTP 4. neurological symptoms → usually TTP 5. renal failure → usually HUS |
Current p485
|
|
|
Define somnolence.
|
drowsiness
|
|
|
|
What labs indicate microangiopathic hemolytic anemia?
|
CBC → anemia
blood smear → schistocytes RETIC → high indirect BILI → high LD → high DC → negative |
|
|
|
What is direct coombs used for?
|
to determine if hemolytic anemia due to antibodies attached to RBCs
|
|
|
|
Define oliguria.
|
low urine output
|
|
|
|
How do you differentiate TTP and HUS?
|
Both:
thrombocytopenia microangiopathic hemolytic anemia TTP: usually affects adults idiopathic fever neurological symptoms HUS: usually affects children often caused by E. coli renal failure stool culture positive for E. coli |
Current p485
|
|
|
What is the difference between plasmapheresis and plasma exchange?
|
plasmapheresis → involved removal of blood, filtration of plasma, and return of patient's plasma
plasma exchange → removal of blood, filtration of plasma, and return of donor plasma |
|
|
|
What is the prognosis of HUS?
|
33% require dialysis
9% develop end stage renal disease 5-15% mortality rate |
|
|
|
What does HUS stand for?
|
hemolytic uremic syndrome
|
|
|
|
What is heparin-induced thrombocytopenia (HIT)?
|
disorder characterized by heparin administration followed by thrombosis and thrombocytopenia
|
Current p486
|
|
|
What is the etiology of HIT?
|
administration of heparin → previous formation of IgG Ab to heparin-platelet factor 4 complexes (i.e. exposed to heparin within last 100 days) → antibodies bind and activate platelets → thrombocytopenia + pro-thrombotic state
|
Current p486
|
|
|
What is the clinical presentation of HIT?
|
onset 5-10 days following heparin therapy
thrombosis bleeding uncommon |
Current p486
|
|
|
What is the diagnostic work-up of HIT?
|
CBC → thrombocytopenia (>50% decline)
PF4-heparin ELISA → positive |
Current p486
|
|
|
What is the management of HIT?
|
1. hospitalize
2. d/c heparin 3. administer direct thrombin inhibitor → argatroban or lepirudin until platelets >100,000/mcL 4. ultrasound for thrombosis 5. warfarin after successful rise in platelets with INR goal of 2.0-3.0 6. if no thrombosis → warfarin x 30 days 7. if thrombosis → warfarin x 3 months 8. avoid heparin unless antibodies eradicated |
Current p486
|
|
|
What are the complications of HIT?
|
thrombosis
|
Current p486
|
|
|
What drugs can cause drug-induced platelet defects?
|
alcohol
omega-3 fatty acids salicylates → aspirin NSAIDs → ibuprofen antibiotics → high dose penicillin, nafcillin, ticarcillin, cephalothin, moxalactam SSRIs dipyridamole thienopryidines glycoprotein IIb/IIIa inhibitors |
Current p490
|
|
|
What is the management of drug-induced platelet defects?
|
1. d/c offending drugs
2. if significant bleeding → platelet transfusion |
Current p490
|
|
|
Define hemarthrosis.
|
bleeding in joints
|
|
|
|
Define arthropathy.
|
disease of the joint
|
|
|
|
What is the clinical presentation of von Willebrand disease?
|
if type 1 → mild to moderate platelet-type bleeding → mucocutaneous bleeding
if type 2 → moderate to severe bleeding |
|
|
|
List drugs that alter coagulation.
|
coagulants:
vitamin K alcohol anticoagulants: alcohol vitamin K angonists → warfarin anti-thrombin → heparin, LMWH, direct thrombin inhibitors antiplatelets → aspirin, thienopyridines (ticlopidine, clopidogrel) |
|
|
|
What are 3 broad causes of bleeding?
|
1. trauma or damage to blood vessels
2. deficienciey in clotting factors 3. physiological disorders of platelets |
Interpreting Laboratory Data p363
|
|
|
Where are platelets found?
|
2/3 found in circulation
1/3 in spleen |
Interpreting Laboratory Data p363
|
|
|
_, _, and _ destroy aging platelets.
|
spleen, liver, and bone marrow
|
Interpreting Laboratory Data p363
|
|
|
What is the lifespan of platelets?
|
8-12 days
|
Interpreting Laboratory Data p363
|
|
|
What is the lifespan of transfused platelets?
|
4-5 days
|
Interpreting Laboratory Data p363
|
|
|
Describe the formation of a platelet plug.
|
vascular injury → collagen exposure → platelet adhesion → platelet aggregation → platelet plug + fibrin → stabilized platelet/fibrin plug
|
Interpreting Laboratory Data p364
|
|
|
How does aspirin work?
|
2. inhibits cyclooxygenase (COX)
2. COX normally catalyzes conversion of arachidonic acid into prostaglandins 3. COX-1 associated with prostaglandins required for synthesis of protective gastric mucous and proper blood flow in kidneys 4. COX-2 associated with prostaglandins for pain, fever, and inflammation |
|
|
|
List factors that promote coagulation.
|
low blood flow or venous stasis → activated clotting factors not cleared
antithrombin, protein C, protein S deficiencies pregnancy immobilization obesity malignancy estrogen therapy |
Interpreting Laboratory Data p367
|
|
|
Warfarin + protein C deficiency may cause?
|
skin necrosis
*avoided by prior anticoagulation with heparin before beginning warfarin |
Interpreting Laboratory Data p366
|
|
|
What is fibrinolysis?
|
process where formed thrombi are lysed to prevent excess clot formation and vascular occlusion
|
Interpreting Laboratory Data p366
|
|
|
Explain the process of fibrinolysis.
|
1. tpA catalyzes plasminogen → plasmin
2. plasmin catalyzes fibrin → fibrin degradation products (FDPs) |
Interpreting Laboratory Data p367
|
|
|
What is the normal range for platelet count?
|
140,000-440,000/mcL
|
Interpreting Laboratory Data p367
|
|
|
What is the ddx for thrombocythemia (>800,000/mcL)?
|
essential thrombocythemia
polycythemia vera CML myelofibrosis |
Interpreting Laboratory Data p366
|
|
|
Risk of spontaneous bleeding occurs when platelets are _?
|
<20,000/mcL
|
Interpreting Laboratory Data p367
|
|
|
What is the normal range for PT?
|
10-13 sec
|
Interpreting Laboratory Data p367
|
|
|
What is the ddx for prolonged PT?
|
factor VII, X, V, II, I deficiencies
vitamin K deficiency liver disease DIC warfarin |
Interpreting Laboratory Data p367
|
|
|
What is the ddx for prolonged PTT?
|
factor XII, XI, IX, VIII, X, V, II, I deficiencies
vitamin K deficiency lupus anticoagulant liver disease DIC heparin warfarin |
|
|
|
If on warfarin therapy, and you reduce the vitamin K in your diet, what may happen?
|
INR may increase
|
Interpreting Laboratory Data p377
|
|
|
What are the indications for prescribing vitamin K?
|
vitamin K deficiency:
inability to synthesize vitamin K → liver disease malabsorption → inflammatory bowel diseases anticoagulant use bulemia abdominal surgery hemorrhagic disease of the newborn |
|
|
|
What foods contain vitamin K?
|
leafy green vegetables
broccoli |
|
|
|
What are the indications for prescribing EPO?
|
anemia related to:
chronic renal failure zidovudine therapy for HIV chemotherapy for metastatic cancer (nonmyeloid malignancies) |
Drug Handbook p531
|
|
|
What are the side effects of ferrous sulfate therapy?
|
GI upset
constipation |
|
|
|
What should you prescribe if ferrous sulfate is not well tolerated?
|
ferrous bi-glycinate chelate → causes less GI upset and constipation
|
|
|
|
What are the indications for prescribing ferrous bi-glycinate chelate?
|
iron deficiency anemia
|
|
|
|
What are the indications for prescribing heparin?
|
prophylaxis and treatment of thromboembolic disorders
|
Drug Handbook p722
|
|
|
What is the management of venous thrombosis?
|
1. LMWH x 5 days
2. warfarin, start at 5mg then adjust accordingly -if thrombus → 3 months -if embolism → 6 months -if recurrent embolism → 12 months 3. monitor INR twice weekly during initiation then monthly with goal 2.0-3.0 |
|
|
|
What are the indications for LMWH therapy?
|
prophylaxis if increased risk of thromboembolism
DVT PE |
|
|
|
What are the indication for warfarin therapy?
|
thromboembolitic disorders:
DVT PE embolitic complications due to: atrial fibrillation cardiac valve replacement prophylaxis following: DVT PE MI |
Drug Handbook p1570
|
|
|
What are the indications for prescribing aspirin?
|
1. mild to moderate fever, inflammation, or pain
2. prophylaxis or treatment of MI, TIA, CVA 3. RA, OA, gout 4. revascularization procedures → CABG, PTCA, stent implantation |
|
|
|
What are the indications for prescribing clopidogrel (Plavix)?
|
1. prophylaxis or treatment of unstable angina, MI, CVA
2. peripheral artery disease |
|
|
|
What are the indications for prescribing ticlodipine?
|
CVA precursors or CVA
*should only be prescribed if aspirin therapy failed or aspirin intolerance present due to high risk of life-threatening hematologic disorders |
Drug Therapy p1468
|
|
|
What type of anemia is folate deficiency anemia?
|
macrocytic megaloblastic
|
|
|
|
What is the clinical presentation of folate deficiency anemia?
|
pallor
mucosal changes → glossitis, diarrhea, anorexia |
|
|
|
What is the diagnostic workup of folate deficiency anemia?
|
CBC reveals ↓ RBCs + ↑ MCV
normal B12 ↓ folate ↓ homocysteine normal methylmelonic acid |
|
|
|
What is the management of folate deficiency anemia?
|
folate supplementation → 1mg PO daily x 2 months
no referral necessary |
|
|
|
Where is folate absorped in the GI tract?
|
entire GI tract
|
|
|
|
How much folate is required daily?
|
50-100 mcg
|
|
|
|
How much folate is stored in the body and how long does that store last?
|
5000 mcg
lasts 2-3 months |
|
|
|
Describe the clotting cascade.
|
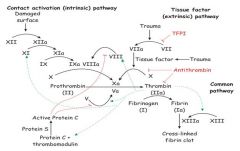
|
|
|
|
What is the ddx for anemia caused by blood loss?
|
hematuria
GI bleed menorrhagia abnormal uterine bleeding blood donation |
|
|
|
What is the ddx for anemia caused by increased destruction of RBCs?
|
INTRINSIC HEMOLYSIS:
1. membrane – hereditary spherocytosis, elliptocytosis 2. hemoglobin – sickle cell disease, unstable hemoglobin 3. glycolysis – pyruvate kinase deficiency 4. oxidation – G6PD deficiency EXTRINSIC HEMOLYSIS: 1. immune – cold antibody, warm antibody 2. microangiopathic – thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, mechanical cardiac valve, paravalvular leak 2. infection – clostridial 3. hypersplenism |
|
|
|
What is the ddx for anemia caused by decreased production of RBCs?
|
1. hemoglobin synthesis – iron deficiency, thalassemia, anemia of chronic disease
2. DNA synthesis – B12 deficiency, folate deficiency 3. stem cells – aplastic anemia, myeloproliferative leukemia 4. bone marrow – carcinoma, lymphoma 5. pure red cell aplasia |
|
|
|
Normocytic anemia indicates?
|
anemia of chronic disease
|
|
|
|
When is parenteral iron indicated for iron deficiency anemia?
|
GI disesae (IBD)
persistent blood loss oral iron not tolerated refractory to oral iron |
|
|
|
How is parenteral iron administered?
|
prescribe sodium ferric gluconate
prescribe 1mg for each mL of RBCs below normal infuse via IV over 4-6 hours monitor for anaphylaxis |
|
|
|
Which clotting factors are dependent on vitamin K for synthesis?
|
II, VII, IX, X
|
|